Desbloquear información biológica a partir de datos genómicos unicelulares complejos se ha vuelto más fácil y preciso gracias a la innovadora herramienta scLENS desarrollada por el Grupo de Matemáticas Biomédicas dentro del Centro de Ciencias Matemáticas y Computacionales del IBS, dirigido por el investigador jefe Kim Jae Kyoung, quien también es Profesor en KAIST. Esto representa un importante avance en el campo de la transcriptómica unicelular.
La investigación se publica en la revista Nature Communications. .
El análisis genómico unicelular es una técnica avanzada que mide la expresión genética a nivel de célula individual, revelando cambios e interacciones celulares que no son observables con los métodos tradicionales de análisis genómico. Cuando se aplica a tejidos cancerosos, este análisis puede delinear la composición de diversos tipos de células dentro de un tumor, proporcionando información sobre cómo progresa el cáncer e identificando genes clave involucrados durante cada etapa de progresión.
A pesar del inmenso potencial del análisis genómico unicelular, manejar la gran cantidad de datos que genera siempre ha sido un desafío. La cantidad de datos cubre la expresión de decenas de miles de genes en cientos o miles de células individuales. Esto no sólo da como resultado grandes conjuntos de datos, sino que también introduce distorsiones relacionadas con el ruido, que surgen en parte debido a las limitaciones actuales de medición.
El autor correspondiente, Kim Jae Kyoung, destacó:"Ha habido un avance notable en las tecnologías experimentales para analizar transcriptomas unicelulares durante la última década. Sin embargo, debido a las limitaciones en los métodos de análisis de datos, ha habido dificultades para utilizar plenamente los datos valiosos obtenidos a través de mucho costo y tiempo."
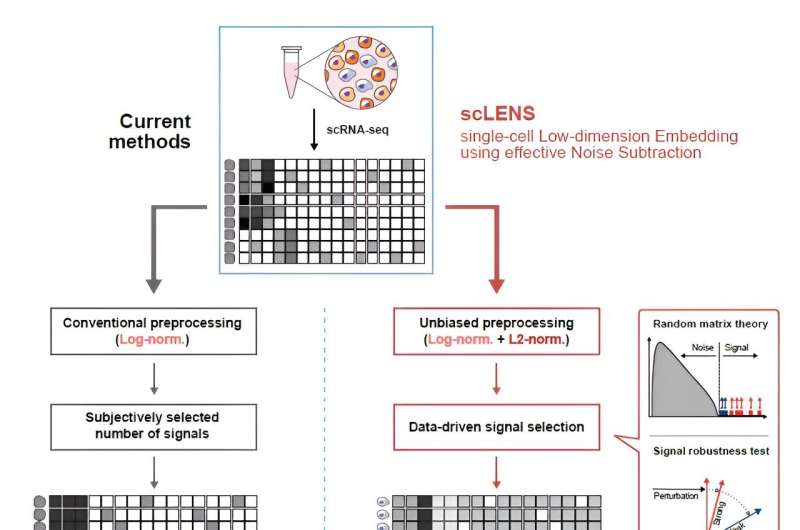
A lo largo de los años, los investigadores han desarrollado numerosos métodos de análisis para discernir las señales biológicas de este ruido. Sin embargo, la precisión de estos métodos no ha sido satisfactoria. Una cuestión crítica es que la determinación de los umbrales de señal y ruido a menudo depende de decisiones subjetivas de los usuarios.
La herramienta scLENS recientemente desarrollada aprovecha la teoría de matrices aleatorias y la prueba de robustez de la señal para diferenciar automáticamente las señales del ruido sin depender de la entrada subjetiva del usuario.
El primer autor, Kim Hyun, afirmó:"Anteriormente, los usuarios tenían que decidir arbitrariamente el umbral de señal y ruido, lo que comprometía la reproducibilidad de los resultados del análisis e introducía subjetividad. scLENS elimina este problema al detectar automáticamente señales utilizando solo la estructura inherente de los datos".
Durante el desarrollo de scLENS, los investigadores identificaron las razones fundamentales de las imprecisiones en los métodos de análisis existentes. Descubrieron que los métodos de preprocesamiento de datos comúnmente utilizados distorsionan tanto las señales biológicas como el ruido. El nuevo enfoque de preprocesamiento que ofrece scLENS está libre de este tipo de distorsiones.
Al resolver problemas relacionados con el umbral de ruido determinado por la elección subjetiva del usuario y la distorsión de la señal en el preprocesamiento de datos convencional, scLENS supera significativamente a los métodos existentes en precisión. Además, scLENS automatiza el laborioso proceso de selección de la dimensión de la señal, lo que permite a los investigadores extraer señales biológicas de forma cómoda y automática.
Ci Kim añadió:"scLENS resuelve problemas importantes en el análisis de datos de transcriptomas unicelulares, mejorando sustancialmente la precisión y la eficiencia durante todo el proceso de análisis. Este es un excelente ejemplo de cómo las teorías matemáticas fundamentales pueden impulsar la innovación en la investigación de las ciencias biológicas, permitiendo a los investigadores obtener más Responde con rapidez y precisión preguntas biológicas y descubre secretos de la vida que antes estaban ocultos."
Más información: Hyun Kim et al, scLENS:detección de señales basada en datos para análisis de datos scRNA-seq imparcial, Nature Communications (2024). DOI:10.1038/s41467-024-47884-3
Información de la revista: Comunicaciones sobre la naturaleza
Proporcionado por el Instituto de Ciencias Básicas














