Uso del aprendizaje automático para ayudar al diseño molecular. Crédito:Wenbo Sun, Avances científicos, doi:10.1126 / sciadv.aay4275
Sintetizar materiales de alto rendimiento para fotovoltaica orgánica (OPV) que conviertan la radiación solar en corriente continua, Los científicos de materiales deben establecer de manera significativa la relación entre las estructuras químicas y sus propiedades fotovoltaicas. En un nuevo estudio sobre Avances de la ciencia , Wenbo Sun y un equipo que incluye investigadores de la Escuela de Energía e Ingeniería Eléctrica, Escuela de Automatización, Ciencias de la Computación, Ingeniería eléctrica y tecnología verde e inteligente, estableció una nueva base de datos de más de 1, 700 materiales de donantes utilizando informes bibliográficos existentes. Utilizaron el aprendizaje supervisado con modelos de aprendizaje automático para construir relaciones estructura-propiedad y materiales de OPV de pantalla rápida utilizando una variedad de entradas para diferentes algoritmos de aprendizaje automático.
Usando huellas digitales moleculares (que codifican una estructura de una molécula en bits binarios) más allá de una longitud de 1000 bits Sun et al. obtuvo una alta precisión de predicción de ML. Verificaron la confiabilidad del enfoque seleccionando 10 materiales de donantes recientemente diseñados para verificar la coherencia entre las predicciones del modelo y los resultados experimentales. Los resultados de ML presentaron una herramienta poderosa para preseleccionar nuevos materiales OPV y acelerar el desarrollo de OPV en la ingeniería de materiales.
Las células fotovoltaicas orgánicas (OPV) pueden facilitar la transformación directa y rentable de la energía solar en electricidad con un rápido crecimiento reciente para superar las tasas de eficiencia de conversión de energía (PCE). La investigación general de la OPV se ha centrado en establecer una relación entre las nuevas estructuras moleculares de la OPV y sus propiedades fotovoltaicas. El proceso tradicional generalmente implica el diseño y síntesis de materiales fotovoltaicos para el ensamblaje / optimización de células fotovoltaicas. Tales enfoques dan como resultado ciclos de investigación que consumen mucho tiempo y requieren un control delicado de la síntesis química y la fabricación de dispositivos. pasos experimentales y purificación. El proceso de desarrollo de OPV existente es lento e ineficiente, con menos de 2000 moléculas donantes de OPV sintetizadas y probadas hasta ahora. Sin embargo, los datos recopilados durante décadas de trabajo de investigación no tienen precio, con valores potenciales que quedan por explorar a fondo para generar materiales OPV de alto rendimiento.
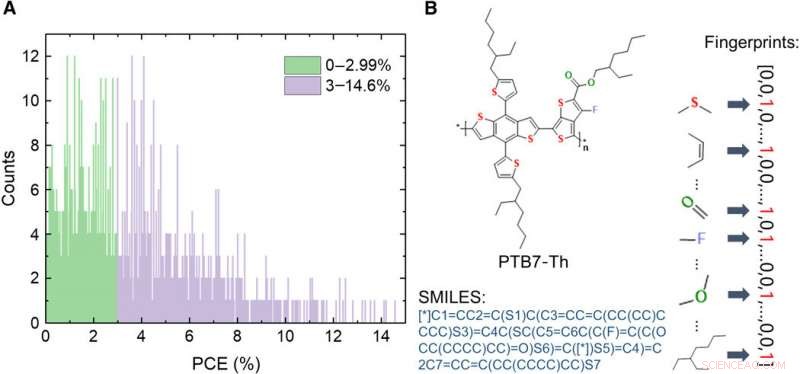
Información sobre la base de datos de materiales de donantes de OPV. (A) Distribución de los valores de PCE de las 1719 moléculas en la base de datos. (B) Esquemas de expresiones de una molécula, incluyendo imagen, sistema simplificado de entrada de línea de entrada molecular (SMILES), y huellas dactilares. Crédito:Science Advances, doi:10.1126 / sciadv.aay4275
Para extraer información útil de los datos, Sun y col. requirió un programa sofisticado para escanear a través de un gran conjunto de datos y extraer relaciones entre las características. Dado que el aprendizaje automático (ML) proporciona herramientas computacionales para aprender y reconocer patrones y relaciones mediante un conjunto de datos de entrenamiento, el equipo utilizó un enfoque basado en datos para habilitar el aprendizaje automático y predecir diversas propiedades de los materiales. El algoritmo ML no tenía que comprender la química o la física detrás de las propiedades de los materiales para realizar las tareas. Métodos similares han predicho recientemente la actividad / propiedades de los materiales con éxito durante el descubrimiento de materiales, desarrollo de fármacos y diseño de materiales. Antes de las aplicaciones de AA, los científicos habían generado la quimioinformática para establecer una caja de herramientas útil.
Los científicos de materiales han explorado recientemente las aplicaciones de ML en el campo de la OPV. En el presente trabajo, Sun y col. estableció una base de datos que contiene 1719 materiales de OPV de donantes probados experimentalmente y recopilados de la literatura. Estudiaron la importancia de la expresión del lenguaje de programación de las moléculas en primer lugar para comprender el rendimiento del aprendizaje automático. Luego probaron varios tipos diferentes de expresiones, incluidas imágenes, Cadenas ASCII, dos tipos de descriptores y siete tipos de huellas dactilares moleculares. Observaron que las predicciones del modelo estaban de acuerdo con los resultados experimentales. Los científicos esperan que el nuevo enfoque acelere en gran medida el desarrollo de materiales semiconductores orgánicos nuevos y altamente eficientes para aplicaciones de investigación de OPV.
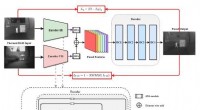
El equipo de investigación primero transformó los datos sin procesar en una representación legible por máquina. Existe una variedad de expresiones para la misma molécula que comprenden información química muy diferente presentada en diferentes niveles abstractos. Usando un conjunto de modelos de AA, Sun y col. exploró diversas expresiones de una molécula comparando su precisión predicha para la eficiencia de conversión de energía (PCE) para obtener una precisión del modelo de aprendizaje profundo del 69,41 por ciento. El desempeño relativamente insatisfactorio se debió al pequeño tamaño de la base de datos. Por ejemplo, anteriormente, cuando el mismo grupo usaba un número mayor de moléculas de hasta 50, 000, la precisión del modelo de aprendizaje profundo superó el 90 por ciento. Para entrenar completamente un modelo de aprendizaje profundo, los investigadores deben implementar una base de datos más grande que contenga millones de muestras.
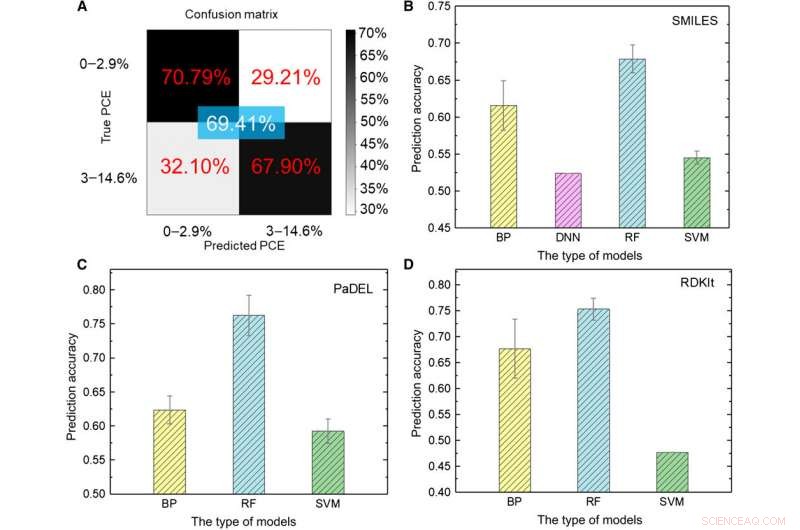
Prueba de resultados de modelos ML. (A) Prueba del modelo de aprendizaje profundo utilizando imágenes como entrada. (B a D) Resultados de las pruebas de diferentes modelos de AA utilizando (B) SONRISAS, (C) PaDEL, y (D) descriptores RDKIt como entrada. Crédito:Science Advances, doi:10.1126 / sciadv.aay4275
Sun y col. solo tenía cientos de moléculas en cada categoría en la actualidad, lo que dificulta que el modelo extraiga suficiente información para una mayor precisión. Si bien es posible ajustar un modelo previamente entrenado para reducir la cantidad de datos necesarios, Todavía se necesitan miles de muestras para lograr un número suficiente de funciones. Esto llevó a la opción de aumentar el tamaño de la base de datos al usar imágenes para expresar moléculas.
Los científicos utilizaron cinco tipos de algoritmos ML supervisados en el estudio, incluyendo (1) red neuronal de retropropagación (BP) (BPNN), (2) red neuronal profunda (DNN), (3) aprendizaje profundo, (4) máquina de vectores de soporte (SVM) y (5) bosque aleatorio (RF). Estos eran algoritmos avanzados, donde BPNN, La DNN y el aprendizaje profundo se basaron en la red artificial neutral (ANN). El código SMILES (sistema simplificado de entrada de línea de entrada molecular) proporcionó otra expresión original de una molécula, que Sun et al. utilizado como entradas para cuatro modelos. Según los resultados, la precisión más alta se aproxima al 67,84 por ciento para el modelo de RF. Como antes, a diferencia del aprendizaje profundo, los cuatro métodos clásicos no pudieron extraer características ocultas. Como un todo, SMILES funcionó peor que las imágenes como descriptores de moléculas para predecir la clase de PCE (eficiencia de conversión de energía) en los datos.
Luego, los investigadores utilizaron descriptores moleculares que pueden describir las propiedades de una molécula utilizando una matriz de números en lugar de la expresión directa de una estructura química. El equipo de investigación utilizó dos tipos de descriptores PaDEL y RDKIt en el estudio. Después de análisis exhaustivos en todos los modelos de AA, un gran tamaño de datos implicaba más descriptores irrelevantes para PCE que afectaban el rendimiento de la RNA. Relativamente, un tamaño de datos pequeño implicaba información química ineficiente para entrenar eficazmente los modelos de aprendizaje automático, cuando se utilizan descriptores moleculares como entrada en enfoques ML, la clave se basó en encontrar descriptores apropiados que se relacionaran directamente con el objeto de destino.
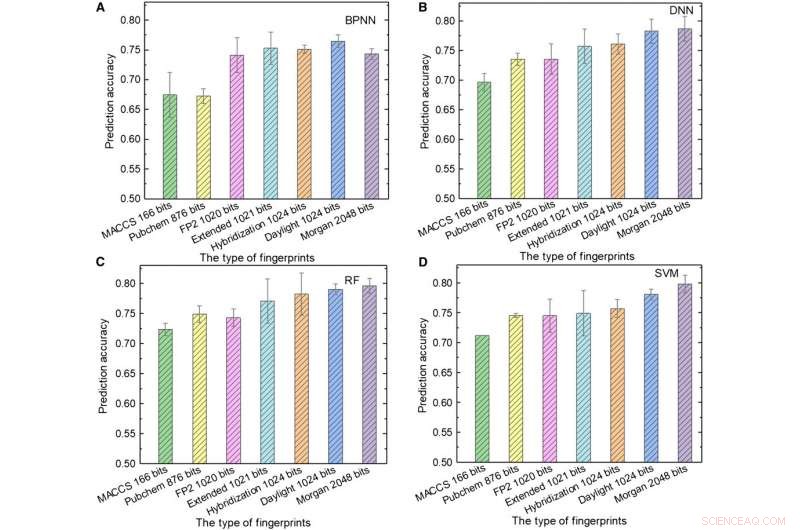
Rendimiento de los modelos ML. (A a D) Los resultados de las pruebas de (A) BPNN, (B) DNN, (C) RF, y (D) SVM usando diferentes tipos de huellas digitales como entrada. Crédito:Science Advances, doi:10.1126 / sciadv.aay4275.
A continuación, el equipo utilizó huellas dactilares moleculares; típicamente diseñado para representar moléculas como objetos matemáticos y originalmente creado para identificar isómeros. Durante el cribado de bases de datos a gran escala, el concepto se representa como una matriz de bits que contienen "1" y "0" para describir la presencia o ausencia de subestructuras o patrones específicos dentro de las moléculas. Sun y col. utilizó siete tipos de huellas dactilares como entradas para entrenar los modelos ML y consideró la influencia de la longitud de la huella dactilar en el rendimiento de predicción de diferentes modelos para obtener diversas huellas dactilares. Por ejemplo, Las huellas dactilares del sistema de acceso molecular (MACCS) contenían 166 bits y eran la entrada más corta y los resultados no eran satisfactorios debido a su información limitada.
Sun y col. mostró la mejor combinación de lenguaje de programación y algoritmo ML obtenida usando huellas dactilares de hibridación de 1024 bits y RF, para lograr una precisión de predicción del 81,76 por ciento; donde las huellas dactilares de hibridación representan estados de hibridación de moléculas SP2. Cuando la longitud de la huella dactilar aumentó de 166 a 1024 bits, el rendimiento de todos los modelos ML mejoró ya que las huellas dactilares más largas incluían más información química.
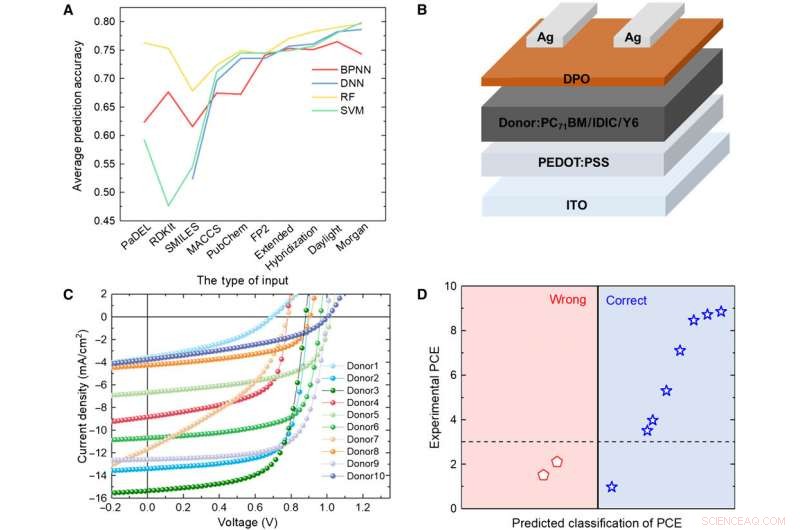
Verificación de modelos ML con experimento. (A) Comparación de los resultados de cuatro modelos diferentes. (B) Diagrama esquemático de la arquitectura celular utilizada en este estudio. (C) Curva J-V de la célula solar con la capa activa utilizando el material donante predicho. (D) Resultados de la predicción frente a datos experimentales para los materiales de donantes previstos con el algoritmo de RF y las huellas dactilares de Daylight. Crédito:Science Advances, doi:10.1126 / sciadv.aay4275.
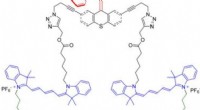
Para probar la confiabilidad de los modelos ML, Sun y col. sintetizó 10 nuevas moléculas donantes de OPV. Luego usó tres huellas digitales representativas para expresar la estructura química de las nuevas moléculas y comparó los resultados predichos por el modelo de RF y los valores experimentales de PCE. El sistema clasificó ocho de las 10 moléculas. Los resultados indicaron el potencial de los materiales sintéticos para aplicaciones OPV con optimización experimental adicional para dos de los nuevos materiales. Un pequeño cambio en la estructura podría causar una gran diferencia en los valores de PCE. En todo alentador, los modelos ML identificaron estas modificaciones menores para facilitar resultados favorables de predicción.
De este modo, Wenbo Sun y sus colegas utilizaron una base de datos bibliográfica sobre materiales de donantes de OPV y una variedad de expresiones de lenguaje de programación (imágenes, Cadenas ASCII, descriptores y huellas dactilares moleculares) para construir modelos ML y predecir la clase de PCE de OPV correspondiente. El equipo demostró un esquema para diseñar materiales de donantes de OPV utilizando enfoques de ML y análisis experimental. Ellos preseleccionaron una gran cantidad de materiales de donantes utilizando el modelo ML para identificar candidatos principales para síntesis y experimentos adicionales. El nuevo trabajo puede acelerar el diseño de nuevos materiales de donantes para acelerar el desarrollo de OPV de PCE altas. El uso de ML junto con experimentos avanzará en el descubrimiento de materiales.
© 2019 Science X Network