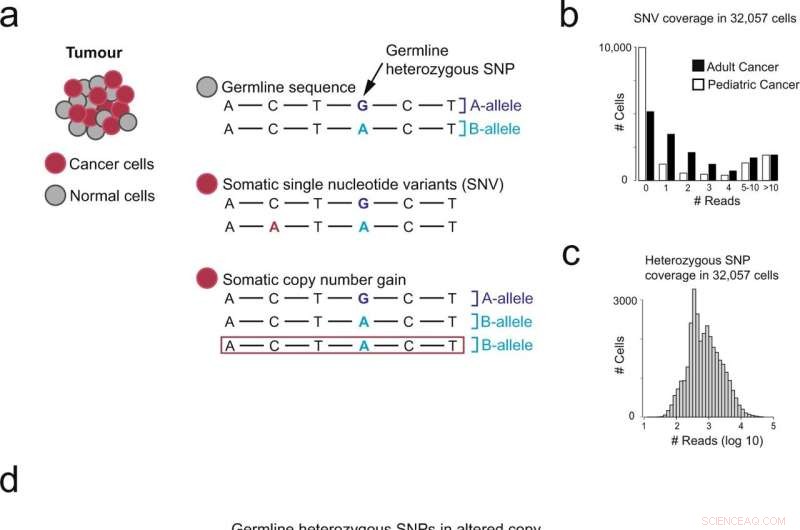
Descripción general de los diferentes enfoques para identificar las células derivadas del cáncer. a Cambios genómicos presentes en los genomas del cáncer. b Número de células (eje y) con N lecturas que cubren mutaciones puntuales (eje x), separadas por carga de mutación baja (neuroblastoma NB) y alta (carcinoma de células renales RCC). c Número de celdas (eje y) con N lecturas que cubren polimorfismos de un solo nucleótido heterocigotos (SNP) (eje x). d Descripción general del uso de cambios alélicos que representan cambios en el número de copias para detectar células cancerosas. Crédito:Biología de las comunicaciones (2022). DOI:10.1038/s42003-022-03808-9. https://www.nature.com/articles/s42003-022-03808-9
Un nuevo método para separar las células cancerosas de las células no cancerosas ha sido desarrollado por investigadores del Instituto Wellcome Sanger, en un impulso para aquellos que trabajan para comprender mejor la biología del cáncer utilizando la secuenciación de ARNm de una sola célula.
El estudio, publicado hoy en Communications Biology , mejora los métodos existentes para identificar qué células de una muestra son cancerosas y proporciona datos cruciales sobre el microambiente de los tumores. El software está disponible abiertamente para que los investigadores de todo el mundo lo apliquen a sus propios datos, mejorando la eficacia de la secuenciación de células individuales para comprender el cáncer.
El análisis de ARNm de células individuales de células cancerosas es una de las técnicas de vanguardia que se utilizan para comprender mejor la biología del cáncer. Los datos generados se pueden usar para tratar de interrumpir los cánceres con medicamentos o averiguar cómo surgen los cánceres en primer lugar.
Un paso fundamental en este proceso es separar las células cancerosas de las no cancerosas, pero no siempre es una tarea fácil. Además de los muchos tipos de cáncer, también habrá diferencias moleculares entre las células cancerosas del mismo tipo dentro de un solo tumor.
Actualmente, el mejor método para hacer esto es medir la expresión génica promedio de las células en la muestra, y se usa una expresión más alta o más baja para distinguir las células cancerosas de las células sanas. Pero este método puede llevar a conclusiones falsas.
En este nuevo estudio, los investigadores del Instituto Wellcome Sanger realizaron la secuenciación del genoma completo y la secuenciación del ARNm de una sola célula en muestras recolectadas por el Great Ormond Street Hospital (GOSH).
Al localizar desequilibrios de alelos en estos datos, que indican cambios en el número de copias en el genoma, el equipo pudo identificar las células cancerosas de forma más fiable que con los métodos anteriores. Este enfoque será principalmente útil para validar nuevos tipos de células cancerosas y comprender mejor el microambiente del tejido tumoral.
"Poder saber cómo el transcriptoma es diferente en células con genomas aberrantes, como los que se encuentran en los cánceres, es un conocimiento valioso y ampliará las preguntas que podemos responder usando la secuenciación de una sola célula", dice el Dr. Matt Young.
El método, llamado alleleIntegrator, está disponible como un paquete de software para que lo utilicen investigadores de todo el mundo. La estimación del nivel de ARNm total específico del tumor predice los resultados del cáncer