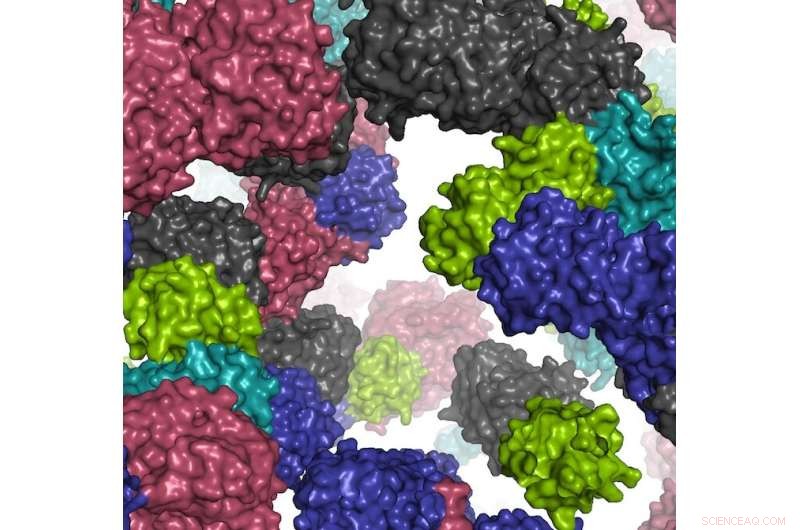
Un fragmento del entorno celular simulado. Crédito:Ilya Vakser
Un informe histórico de la Universidad de Kansas que aparece esta semana en las Proceedings of the National Academy of Sciences propone una nueva técnica para modelar vida molecular con computadoras.
Según el autor principal Ilya Vakser, director del Programa de Biología Computacional y Centro de Biología Computacional y profesor de biociencias moleculares en KU, la investigación sobre el modelado por computadora de los procesos de la vida es un paso importante hacia la creación de una simulación funcional de una célula viva con resolución atómica. . El avance promete nuevos conocimientos sobre la biología fundamental de una célula, así como un tratamiento más rápido y preciso de las enfermedades humanas.
"Es unas decenas o cientos de miles de veces más rápido que las técnicas de resolución atómica existentes", dijo Vakser. "Esto brinda oportunidades sin precedentes para caracterizar los mecanismos fisiológicos que ahora están mucho más allá del alcance del modelado computacional, para obtener información sobre los mecanismos celulares y utilizar este conocimiento para mejorar nuestra capacidad para tratar enfermedades".
Hasta ahora, un obstáculo importante para el modelado de células por computadora ha sido cómo abordar las proteínas y sus interacciones que se encuentran en el corazón de los procesos celulares. Hasta la fecha, las técnicas establecidas para modelar las interacciones de proteínas han dependido del "acoplamiento de proteínas" o de la "simulación molecular".
Según los investigadores, ambos enfoques tienen ventajas y desventajas. Si bien los algoritmos de acoplamiento de proteínas son excelentes para muestrear coordenadas espaciales, no tienen en cuenta la "coordenada de tiempo" o la dinámica de las interacciones de proteínas. Por el contrario, las simulaciones moleculares modelan bien la dinámica, pero estas simulaciones son demasiado lentas o de baja resolución.
"Nuestro estudio de prueba de concepto une las dos metodologías de modelado, desarrollando un enfoque que puede alcanzar escalas de tiempo de simulación sin precedentes en una resolución de todos los átomos", escribieron los autores.
Los colaboradores de Vakser en el artículo fueron Sergei Grudinin de la Universidad de Grenoble Alpes en Francia; Eric Deeds de la Universidad de California-Los Ángeles; Nathan Jenkins, estudiante de doctorado de KU, y Petras Kundrotas, profesor asistente de investigación del Programa de Biología Computacional de KU.
Después de conceptualizar la mejor manera de combinar las ventajas de los dos enfoques de modelado de proteínas, el equipo desarrolló y codificó un algoritmo para impulsar la nueva simulación.
"El desafío más difícil fue desarrollar el algoritmo que refleja adecuadamente la idea básica simple del enfoque", dijo Vakser.
Pero una vez que lograron ese avance, pudieron comenzar a validar el nuevo procedimiento.
"El paradigma fue fácil:un golpe de claridad", dijo Vakser.
"Los enfoques de simulación existentes pasan la mayor parte del tiempo de cómputo viajando en áreas del sistema de baja probabilidad o alta energía. Todos sabemos dónde están estas áreas. En cambio, la idea era muestrear, o viajar, solo en el alto -probabilidad, áreas de baja energía y omitir las de baja probabilidad mediante la estimación de las tasas de transición entre los estados de alta probabilidad. El paradigma es tan antiguo como el propio modelado biomolecular y ha sido ampliamente utilizado desde los albores de la era del modelado. hace décadas".
Pero Vakser dijo que hasta el nuevo artículo de su equipo, el enfoque no se había aplicado a la cinética de las interacciones de proteínas en el entorno celular, el foco de su estudio.
"Debido a que hay muchos menos estados de alta probabilidad que los de baja probabilidad, eso nos dio una gran ganancia en la velocidad de cálculo, de decenas a cientos de miles de veces", dijo Vakser. "Esto se hizo sin pérdida aparente de precisión. Se puede argumentar que se ganó precisión, porque el protocolo de simulación se basa en las técnicas de 'acoplamiento', que están diseñadas específicamente para caracterizar ensamblajes de proteínas".
El investigador de KU dijo que su método de simulación celular podría implementarse para investigar la salud humana y tratar enfermedades con un nuevo nivel de precisión.
"El enfoque se puede utilizar para estudiar las vías moleculares que subyacen a los mecanismos de la enfermedad", dijo Vakser. "Se puede usar para determinar los efectos nocivos de las mutaciones genéticas por los patrones modificados de las asociaciones de proteínas:las mutaciones genéticas provocan cambios en la estructura de las proteínas, lo que a su vez afecta la asociación de proteínas. O podría usarse para identificar objetivos para el diseño de fármacos mediante detectar elementos críticos en los patrones de asociación de proteínas".
Según Vakser, la nueva técnica de simulación ofrece muchas vías de investigación prometedoras para explorar en el futuro.
“Entre ellos están adaptando el enfoque a las interacciones de proteínas con ácidos nucleicos, ARN y ADN”, dijo. "Además, nos gustaría tener en cuenta la flexibilidad de las formas moleculares, correlacionarlas con el espectro de estudios experimentales del entorno celular que se desarrolla rápidamente y aplicar el procedimiento a un modelo de una célula real, con sus componentes moleculares reales empaquetados". La ciencia en la cúspide de la comprensión "transformacional" de la vida a través del modelado celular, dicen los investigadores