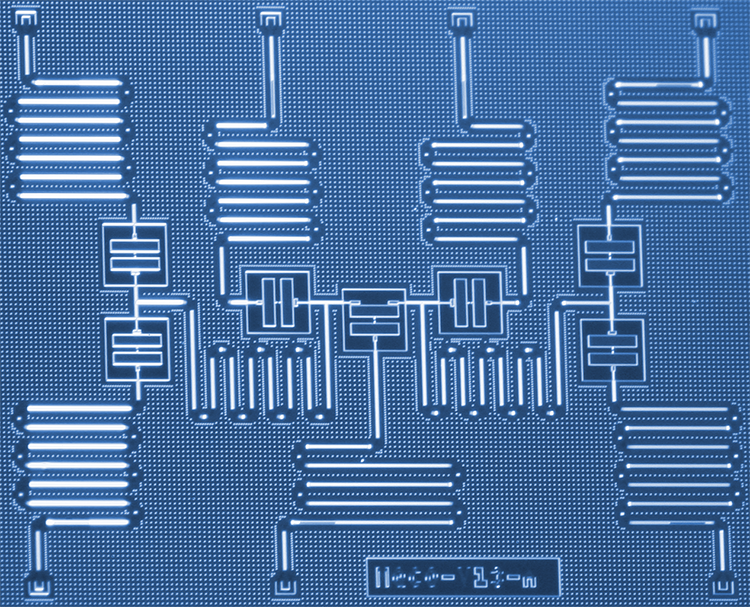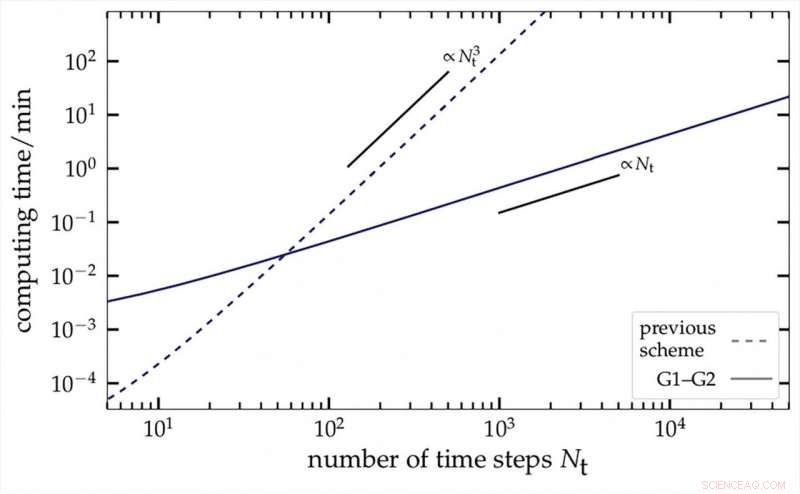
Tiempo de cálculo requerido para el nuevo método G1-G2 (línea continua) en función de la duración del proceso, en comparación con el método tradicional (escala logarítmica). Crédito:Niclas Schlünzen, AG Bonitz
Cómo se comporta un electrón en un átomo, o cómo se mueve en un sólido, se puede predecir con precisión con las ecuaciones de la mecánica cuántica. Estos cálculos teóricos concuerdan completamente con los resultados obtenidos de los experimentos. Pero los sistemas cuánticos complejos, que contienen muchos electrones o partículas elementales, como moléculas, sólidos o núcleos atómicos:actualmente no se puede describir con exactitud, incluso con las computadoras más potentes disponibles en la actualidad. Las ecuaciones matemáticas subyacentes son demasiado complejas, y los requisitos computacionales son demasiado grandes. Un equipo dirigido por el profesor Michael Bonitz del Instituto de Física Teórica y Astrofísica de la Universidad de Kiel (CAU) ha logrado desarrollar un método de simulación, que permite cálculos de mecánica cuántica hasta alrededor de 10, 000 veces más rápido de lo que era posible anteriormente. Han publicado sus hallazgos en el número actual de la reconocida revista científica Cartas de revisión física .
Incluso con computadoras extremadamente potentes, las simulaciones cuánticas tardan demasiado
El nuevo procedimiento de los investigadores de Kiel se basa en una de las técnicas de simulación más potentes y versátiles de la actualidad para sistemas de muchos cuerpos de la mecánica cuántica. Utiliza el método de las llamadas funciones de Green de desequilibrio:esto permite describir movimientos e interacciones complejas de electrones con una precisión muy alta. incluso durante un período prolongado. Sin embargo, Hasta la fecha, este método requiere una gran cantidad de computadoras:para predecir el desarrollo del sistema cuántico durante un período diez veces más largo, una computadora requiere mil veces más tiempo de procesamiento.
Con el truco matemático de introducir una variable auxiliar adicional, Los físicos de la CAU ahora han logrado reformular las ecuaciones primarias de las funciones de Green de desequilibrio de manera que el tiempo de cálculo solo aumenta linealmente con la duración del proceso. Por lo tanto, un período de predicción diez veces más largo solo requiere diez veces más tiempo de cálculo. En comparación con los métodos utilizados anteriormente, los físicos lograron un factor de aceleración de aproximadamente 10, 000. Este factor aumenta aún más para procesos más largos. Dado que el nuevo enfoque combina dos funciones verdes por primera vez, se llama "método G1-G2".
El desarrollo temporal de las propiedades de los materiales es predecible por primera vez.
El nuevo modelo de cálculo del equipo de investigación de Kiel no solo ahorra un costoso tiempo de cálculo, pero también permite simulaciones, que antes eran completamente imposibles. "Nos sorprendió que esta espectacular aceleración también se pueda demostrar en aplicaciones prácticas, "explicó Bonitz. Por ejemplo, Ahora es posible predecir cómo se desarrollan ciertas propiedades y efectos en materiales como los semiconductores durante un período de tiempo prolongado. Bonitz está convencido:"El nuevo método de simulación es aplicable en numerosas áreas de la teoría cuántica de muchos cuerpos, y permitirá predicciones cualitativamente nuevas, como sobre el comportamiento de los átomos, moléculas, plasmas densos y sólidos después de la excitación por radiación láser intensa ".