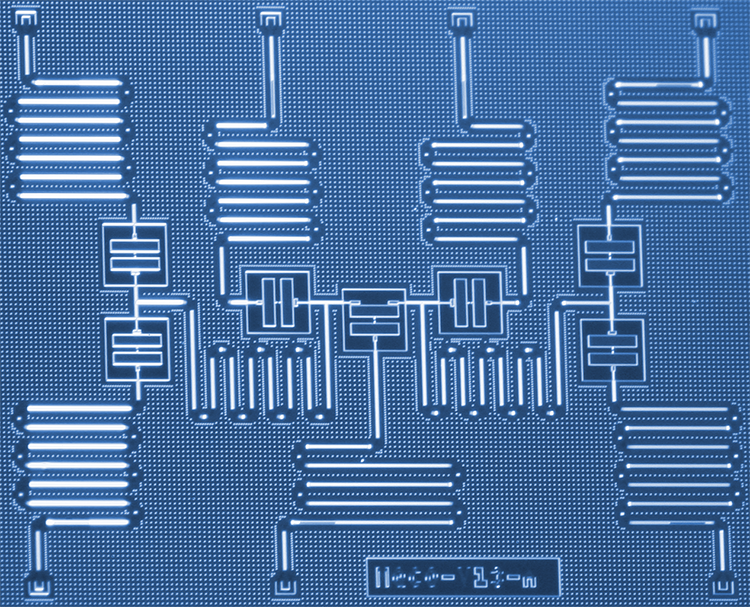
Los científicos de IBM han desarrollado un nuevo enfoque para simular moléculas en una computadora cuántica que algún día podría ayudar a revolucionar la química y la ciencia de los materiales. Los científicos utilizaron con éxito seis qubits en un procesador cuántico de siete qubits especialmente diseñado para abordar el problema de la estructura molecular del hidruro de berilio (BeH2), la molécula más grande simulada en una computadora cuántica hasta la fecha. Los resultados demuestran un camino de exploración de sistemas cuánticos a corto plazo para mejorar nuestra comprensión de reacciones químicas complejas que podrían conducir a aplicaciones prácticas. Crédito:Kandala et al .; Naturaleza
Simular moléculas en computadoras cuánticas ahora es mucho más fácil con el hardware cuántico superconductor de IBM. En un artículo de investigación reciente publicado en Naturaleza , Eigensolver cuántico variacional de hardware eficiente para moléculas pequeñas e imanes cuánticos, implementamos un nuevo algoritmo cuántico capaz de calcular de manera eficiente el estado de menor energía de las moléculas pequeñas. Al mapear la estructura electrónica de los orbitales moleculares en un subconjunto de nuestro procesador cuántico de siete qubit especialmente diseñado, estudiamos moléculas previamente inexploradas con computadoras cuánticas, incluyendo hidruro de litio (LiH) e hidruro de berilio (BeH2). La codificación particular de orbitales a qubits estudiada en este trabajo se puede utilizar para simplificar las simulaciones de moléculas aún más grandes y esperamos la oportunidad de explorar simulaciones más grandes en el futuro. cuando el poder computacional cuántico (o "volumen cuántico") de los sistemas IBM Q ha aumentado.
Si bien BeH2 es la molécula más grande jamás simulada por una computadora cuántica hasta la fecha, el modelo considerado de la molécula en sí es todavía lo suficientemente simple como para que las computadoras clásicas lo simulen con exactitud. Esto lo convirtió en un caso de prueba para superar los límites de lo que podía lograr nuestro procesador de siete qubit, ampliar nuestra comprensión de los requisitos para mejorar la precisión de nuestras simulaciones cuánticas, y sentar los elementos fundamentales necesarios para explorar tales estudios de energía molecular.
Las mejores simulaciones de moléculas actuales se ejecutan en computadoras clásicas que utilizan métodos aproximados complejos para estimar la energía más baja de un hamiltoniano molecular. Un "hamiltoniano" es un operador de energía mecánica cuántica que describe las interacciones entre todos los orbitales de electrones y los núcleos de los átomos constituyentes. El estado de "energía más baja" del hamiltoniano molecular dicta la estructura de la molécula y cómo interactuará con otras moléculas. Esta información es fundamental para que los químicos diseñen nuevas moléculas, reacciones, y procesos químicos para aplicaciones industriales.
Qubit:Orbital
Aunque nuestro procesador cuántico de siete qubits no está completamente corregido a errores ni es tolerante a fallas, los tiempos de coherencia de los qubits individuales duran alrededor de 50 µs. Por lo tanto, es realmente importante utilizar un algoritmo cuántico muy eficiente para aprovechar al máximo nuestra preciosa coherencia cuántica e intentar comprender las estructuras moleculares. El algoritmo tiene que ser eficiente en términos de número de qubits utilizados y número de operaciones cuánticas realizadas.

Aplicación a la química cuántica. C.A, Resultados experimentales (círculos negros rellenos), superficies de energía exactas (líneas de puntos) y diagramas de densidad (sombreado; ver escalas de color) de los resultados de las simulaciones numéricas, para varias distancias interatómicas para H2 (a), LiH (b) y BeH2 (c). Los resultados experimentales y numéricos presentados son para circuitos de profundidad d =1. Las barras de error en los datos experimentales son más pequeñas que el tamaño de los marcadores. Las gráficas de densidad se obtienen a partir de 100 resultados numéricos en cada distancia interatómica. Los recuadros superiores de cada panel resaltan los qubits utilizados para el experimento y las puertas de resonancia cruzada (flechas, etiquetado CRc – t; donde 'c' denota el qubit de control y 't' el qubit objetivo) que constituyen UENT. Los recuadros inferiores son representaciones de la geometría molecular (no a escala). Para las tres moléculas, la desviación de los resultados experimentales de las curvas exactas está bien explicada por las simulaciones estocásticas. Crédito:Kandala et al .; Naturaleza
Nuestro esquema contrasta con los algoritmos de simulación cuántica previamente estudiados, que se centran en adaptar los esquemas clásicos de simulación molecular al hardware cuántico y, al hacerlo, no tienen en cuenta de manera efectiva los gastos generales limitados de los dispositivos cuánticos realistas actuales.
Entonces, en lugar de imponer métodos de computación clásicos al hardware cuántico, hemos invertido el enfoque y preguntamos:¿cómo podemos extraer la máxima potencia computacional cuántica de nuestro procesador de siete qubit?
Nuestra respuesta a esto combina una serie de técnicas eficientes en hardware para atacar el problema:
Con los futuros procesadores cuánticos, que tendrá más volumen cuántico, Podremos explorar el poder de este enfoque de simulación cuántica para moléculas cada vez más complejas que están más allá de las capacidades informáticas clásicas. La capacidad de simular reacciones químicas con precisión, es propicio para los esfuerzos de descubrir nuevos medicamentos, fertilizantes, incluso nuevas fuentes de energía sostenibles.
Los experimentos que detallamos en nuestro artículo no se ejecutaron en nuestros procesadores de cinco y 16 qubit disponibles públicamente en la nube. Pero los desarrolladores y usuarios de la experiencia IBM Q ahora pueden acceder a los cuadernos Jupyter de química cuántica en el repositorio Github de QISKit. En el sistema de cinco qubit, los usuarios pueden explorar la simulación de energía del estado fundamental para las moléculas pequeñas de hidrógeno y LiH. Los portátiles para moléculas más grandes están disponibles para aquellos con acceso beta al procesador actualizado de 16 qubits.