Amoníaco (NH3 ) es una molécula importante con muchas aplicaciones. Producto final del famoso proceso Haber-Bosch, se sintetiza comúnmente para capturar nitrógeno para fertilizantes y se utiliza para refrigeración, productos de limpieza y producción de productos farmacéuticos. Recientemente, esta modesta molécula también ha atraído interés como recurso potencial para abordar uno de los desafíos más apremiantes de la actualidad:la necesidad de combustibles renovables confiables y abundantes.
El amoníaco es estable y seguro de manipular, es combustible y contiene la mayor fracción de hidrógeno de cualquier molécula, excepto el hidrógeno puro. Estos factores prometen convertirlo en una alternativa viable a los vectores de energía basados en el carbono que están impulsando el cambio climático. Se han comenzado a investigar cómo se podría utilizar el amoníaco para alimentar directamente motores, turbinas de gas y pilas de combustible de hidrógeno, por ejemplo. También se cree que el amoníaco podría usarse para almacenar energía para momentos en que otras energías renovables como la eólica y la solar no puedan satisfacer la demanda.
Se sabe mucho sobre el amoníaco, pero este interés por utilizarlo como combustible ha iniciado la búsqueda de nuevas tecnologías de amoníaco. Esto, a su vez, ha llevado a una mayor necesidad entre los ingenieros químicos de datos precisos que describan las propiedades termodinámicas fundamentales del amoníaco. Dichas propiedades incluyen una amplia variedad de rasgos mensurables, como el equilibrio de fases, la densidad o la capacidad calorífica, por ejemplo, que caracterizan los sistemas físicos y determinan cómo funcionan los procesos químicos. En el caso del amoníaco, a los ingenieros también les gustaría conocer mejor cómo cambian estas propiedades al mezclar amoníaco con otras moléculas. Este conocimiento podría ayudarles a optimizar los procesos y las condiciones operativas.
El Dr. Jadran Vrabec, actualmente director del Instituto de Ciencias de Procesos de la Universidad Técnica de Berlín, ha pasado gran parte de su carrera utilizando la computación de alto rendimiento (HPC) para investigar propiedades termodinámicas a nivel molecular. "Las propiedades termodinámicas están determinadas al 100% por interacciones moleculares", explica. "Y debido a que estas interacciones ocurren tan rápido y a una escala tan pequeña, sólo es posible estudiarlas realizando grandes simulaciones usando supercomputadoras".
En un artículo reciente publicado en el Journal of Chemical &Engineering Data Él y el coautor Erich Mace, de la Universidad Técnica de Berlín, informan sobre los resultados de simulaciones centradas en las propiedades termodinámicas de mezclas que contienen amoníaco. Producidos utilizando la supercomputadora Hawk en el Centro de Computación de Alto Rendimiento de Stuttgart (HLRS), sus resultados agregan datos valiosos que podrían respaldar el desarrollo de nuevas aplicaciones del amoníaco. Los resultados también podrían ayudar a evaluar la precisión de otros datos existentes, garantizando que los ingenieros tengan la mejor información disponible para trabajar con la sustancia.
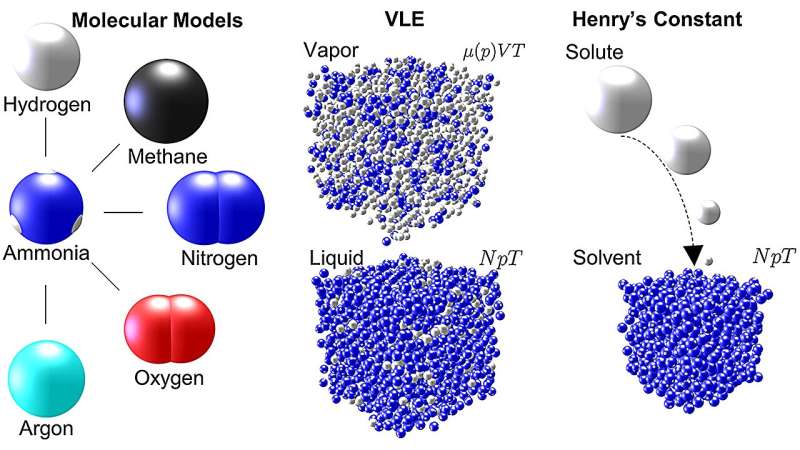
Las simulaciones a gran escala proporcionan información única sobre las propiedades termodinámicas
Vrabec es un usuario desde hace mucho tiempo de los recursos de supercomputación HLRS para dinámica molecular y simulaciones de Monte Carlo. Su enfoque se basa en conceptos de termodinámica que fueron articulados por primera vez por Ludwig Boltzmann en el siglo XIX, pero que su aplicación sólo se volvió práctica en la década de 1950 con la llegada de las primeras computadoras. Desde entonces, el campo ha avanzado en paralelo con el desarrollo de supercomputadoras más grandes y más rápidas, hasta el punto de que las simulaciones de Vrabec ahora rastrean los movimientos individuales y las interacciones de miles de millones o incluso billones de moléculas simultáneamente. Utilizando el software que desarrolló su laboratorio para capturar selectivamente datos de interés, puede luego estudiar las propiedades termodinámicas de las moléculas.
Vrabec utiliza dos códigos de simulación llamados ms2 y ls1, que ha desarrollado y optimizado a lo largo de una larga y fructífera colaboración con los miembros del personal de HLRS, Martin Bernreuther y Christoph Niethammer. En 2019, el equipo incluso estableció un récord mundial para el sistema molecular más grande jamás simulado utilizando métodos de dinámica molecular. Usando ls1, escalaron eficientemente su código a un sistema de 21 billones de átomos en el que se podía rastrear cada molécula individual y sus interacciones con otras moléculas.
En el trabajo reciente sobre amoníaco, Mace y Vrabec realizaron dinámica molecular y simulaciones de Monte Carlo utilizando ms2 para investigar cinco mezclas comúnmente utilizadas que involucran amoníaco en procesos de ingeniería química:argón-amoníaco, metano-amoníaco, hidrógeno-amoníaco, nitrógeno-amoníaco y oxígeno. -amoníaco. Para cada mezcla, las simulaciones generaron datos que describen el equilibrio vapor-líquido (VLE), una medida de la distribución de moléculas en un sistema a través de las fases de vapor o líquido, para un amplio rango de temperaturas y presiones.
En su artículo, Mace y Vrabec señalan que los datos VLE se utilizan a menudo para desarrollar ecuaciones de estado para fluidos industriales; es decir, los datos se pueden utilizar para predecir el estado de la materia en diferentes condiciones físicas debido a cambios de temperatura, presión, volumen o composición. Esta información es esencial para determinar mezclas óptimas y condiciones de trabajo en aplicaciones industriales.
Las simulaciones moleculares de Vrabec son particularmente valiosas porque pueden usarse para investigar una gama de escalas mucho más amplia que la que es posible utilizando enfoques experimentales.
"En nuestras simulaciones hemos medido propiedades termodinámicas incluso con presiones de hasta 50 megapascales. Esto es 500 veces la presión del aire ambiente", comenta Vrabec. "Aunque hace más de un siglo que se recopilan datos sobre las mezclas de amoníaco, su cobertura es sorprendentemente limitada. La razón es que el esfuerzo para medirlas experimentalmente es prohibitivamente enorme. Se necesitarían equipos especiales costosos y su funcionamiento sería peligroso. Simulaciones por computadora, podemos obtener resultados de forma segura y relativamente económica". Sus métodos también proporcionan un nivel de precisión comparable al de los enfoques experimentales en rangos donde hay datos experimentales disponibles.
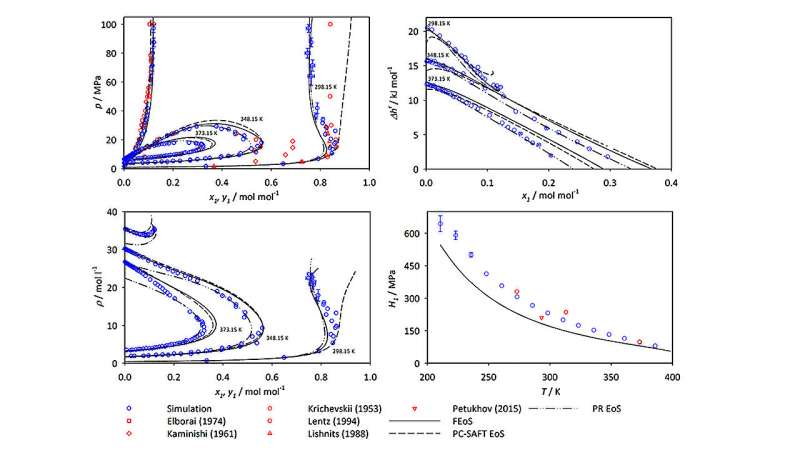
Mejores datos para la investigación del amoníaco
Cuando Mace y Vrabec analizaron los datos de su simulación, demostraron que, aunque el amoníaco es un componente en los cinco sistemas que estudiaron, los gráficos resultantes de los valores VLE parecen dramáticamente diferentes para diferentes mezclas moleculares. Según Vrabec, "el comportamiento de fase de diferentes mezclas está fuertemente determinado por las interacciones entre las moléculas del sistema. Es necesario comprender estas propiedades si está interesado en trabajar con mezclas de amoníaco".
El artículo y sus datos complementarios ofrecen más de 400 nuevos puntos de datos para cada mezcla que estudiaron. Utilizando Hawk, pudieron producir los resultados de cada mezcla en tan solo unos días de tiempo de cálculo. Los resultados serán de particular valor para condiciones extremas y difíciles de estudiar para las cuales hay pocos datos disponibles, y podrían ayudar a los ingenieros a identificar puntos óptimos donde las condiciones serían óptimas para un procesamiento eficiente del amoníaco.
El estudio incluyó tanto nuevos datos de simulación como datos publicados previamente de la literatura científica, lo que permitió a Mace y Vrabec comparar sus resultados con otros conjuntos de datos existentes de valores VLE. En la mayoría de las situaciones, sus resultados se correspondieron estrechamente con los de estudios anteriores. En algunos casos, sin embargo, identificaron divergencias significativas entre sus resultados y las mediciones y predicciones generadas experimentalmente por otros grupos de investigación. Los autores atribuyen estas discrepancias a limitaciones o imprecisiones en los métodos experimentales correspondientes. También sugieren que las fuentes de datos experimentales específicas se deben utilizar con precaución en futuras investigaciones o aplicaciones de ingeniería química.
Vrabec dice que en trabajos recientes se ha centrado principalmente en simular propiedades termodinámicas de sistemas moleculares, generalmente a escala submicrométrica. A pesar de los muchos órdenes de magnitud que se encuentran entre esta escala y el nivel de los procesos observables, existen métodos precisos para traducir estos conocimientos a nivel molecular en predicciones útiles del mundo real.
Sin embargo, a medida que las supercomputadoras crecen, anticipa que también podría ser posible simular no solo propiedades sino también procesos termodinámicos utilizando condiciones límite cercanas a las aplicaciones del mundo real. Un mayor rendimiento de HPC podría producir resultados más precisos sobre fenómenos dinámicos con una mejor relación señal-ruido.
Mientras tanto, sin embargo, los resultados de su equipo demuestran el valor de la dinámica molecular y la simulación Monte Carlo utilizando computación de alto rendimiento, y proporcionarán una nueva comprensión del comportamiento de las fases que los ingenieros pueden utilizar para desarrollar nuevas tecnologías basadas en amoníaco.
Más información: Erich J. Mace et al, Equilibrios de fase fluida a alta presión y constantes de Henry de gases supercríticos en amoníaco, Journal of Chemical &Engineering Data (2023). DOI:10.1021/acs.jced.3c00327
Proporcionado por el Centro Gauss de Supercomputación