Gráficamente abstracto. Crédito:Diario de Biología Molecular (2022). DOI:10.1016/j.jmb.2022.167525
¿Qué genes son específicos para un determinado tipo de célula, es decir, "marcan" su identidad? Con el tamaño cada vez mayor de los conjuntos de datos en la actualidad, responder a esta pregunta suele ser un desafío. A menudo, los genes marcadores son simplemente genes que se han encontrado en poblaciones celulares específicas. Sin embargo, muchos más genes podrían ser característicos de un tipo de célula en particular, pero aún no se han descubierto.
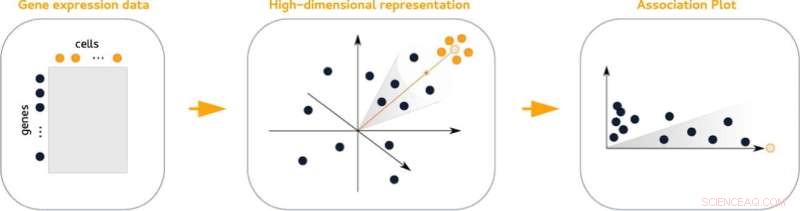
Los diagramas de asociación (APL), un nuevo método estadístico para visualizar la actividad de los genes dentro de un grupo de células, facilitan la búsqueda de sus genes marcadores. Los gráficos comparan la actividad de los genes de un grupo determinado con todos los demás grupos del conjunto de datos. Además, facilitan ver qué genes se comparten con otros grupos.
"Los diagramas de asociación no solo nos permiten identificar nuevos genes marcadores. También funciona al revés:podemos hacer coincidir grupos de identidad desconocida en un conjunto de datos con tipos de células, según una lista proporcionada de genes marcadores", dice Elzbieta Gralinska. del Instituto Max Planck de Genética Molecular de Berlín.
El biotecnólogo trabaja en el equipo de Martin Vingron, que desarrolló la técnica. Los investigadores demostraron la funcionalidad de la técnica en dos conjuntos de datos disponibles públicamente y publicaron los resultados en el Journal of Molecular Biology. . Además, APL se lanzó como un módulo gratuito para el entorno estadístico R. El paquete APL permite a los investigadores inspeccionar visualmente sus datos unicelulares y seleccionar genes individuales con el cursor para obtener más detalles.
Análisis y agrupación de celdas individuales
¿Por qué es necesario identificar genes marcadores en primer lugar? Las modernas tecnologías de secuenciación pueden descifrar moléculas de ARN individuales en células individuales. A partir de una muestra de sangre, por ejemplo, se puede separar cada célula y se puede decodificar una muestra de los ARN de la célula. Estos datos unicelulares representan los genes activos que se transcribieron en moléculas de ARN.
La ventaja:en lugar de preguntarse a qué tipo de célula pertenece un ARN en particular, se puede rastrear hasta su célula de origen. La desventaja:la secuenciación de miles de ARN en cada célula de decenas de miles de células produce cantidades extraordinarias de datos.
Una salida es clasificar las células según su contenido de ARN. "Los datos de una sola celda se componen de una mezcla salvaje de muchos tipos de células diferentes. Estamos interesados en las células del mismo tipo de célula, que deberían comportarse de manera similar", explica Martin Vingron. Por lo tanto, tiene sentido agrupar computacionalmente celdas similares, dice. "Para nosotros, los genes marcadores definen un tipo de célula".
Exploración de grupos de células de forma interactiva
Usando datos disponibles públicamente de glóbulos blancos, el equipo demostró cómo funciona el nuevo algoritmo. Los muchos tipos diferentes de glóbulos blancos, como las células T, las células B o los monocitos, se agrupan en grupos separados. Los investigadores confirmaron genes marcadores conocidos y pudieron demostrar que los parientes cercanos entre las células sanguíneas también comparten una gran similitud en su actividad genética.
"Cada uno de los genes marcadores que encontramos con APL podría haber sido descubierto por al menos otro método existente para la identificación de genes marcadores", dice Gralinska. Pero la ventaja de APL sobre los algoritmos existentes es su representación gráfica de los resultados, dice. "Las herramientas existentes proporcionan largas listas de genes y valores de puntuación. A menudo, los usuarios revisan la lista y se detienen en un punto de corte arbitrario".
Por el contrario, el nuevo método proporciona una forma de visualizar estos genes, hacer clic en cada uno y observar más de cerca su actividad, dice ella. "No solo proporcionamos listas de genes marcadores, sino que permitimos que los usuarios revisen cómo se comportan estos genes", dice el investigador. "Con gráficos de asociación, pueden sumergirse en sus datos para aprender más sobre cada tipo de célula". Además, dice, es muy fácil desglosar la función biológica de los genes más interesantes en un paso posterior a través del análisis de enriquecimiento de términos de Gene Ontology, que es compatible con el software APL, algo que ella considera "una función muy útil".
El modelo matemático subyacente
Los datos de alta dimensión que contienen información sobre la actividad entre genes no se pueden representar visualmente sin pérdida de información. Lo mismo ocurre con los datos agrupados, todo lo cual complica el análisis. "Nuestro truco es que tenemos en cuenta mucho más que solo dos o tres dimensiones, pero finalmente creamos un diagrama de dos dimensiones", dice Gralinska.
Los diagramas de asociación se derivan de una técnica matemática que incrusta simultáneamente genes y células en un espacio común de alta dimensión. La medición de las distancias entre los genes y un grupo de células determinado en este espacio da como resultado pares de valores que reflejan la asociación de un gen con un grupo determinado y brindan información sobre su asociación con otros grupos.
"Una deficiencia de APL es que dependemos de datos preagrupados, lo que significa que tenemos que confiar en otras técnicas para la agrupación", dice Martin Vingron. "Sin embargo, esperamos que nuestro nuevo método encuentre muchos usuarios nuevos. Descubrimos que un proceso visual e interactivo simplemente hace un mejor análisis".