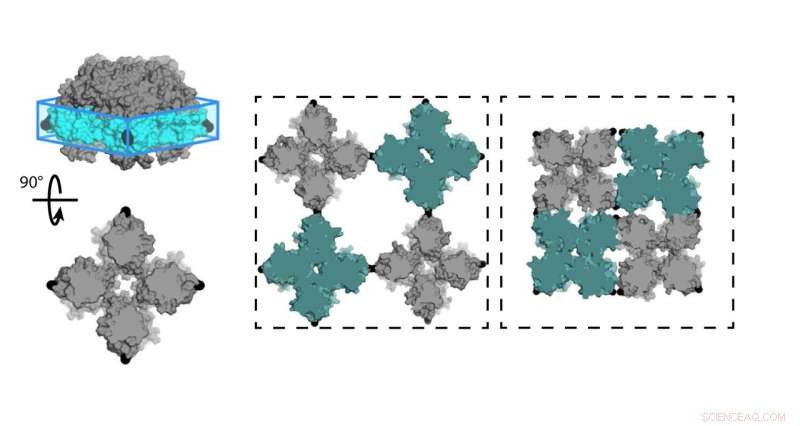
Químicos de la Universidad de California, San Diego (UCSD) diseñó una hoja de proteínas (C98RhuA) que alternan entre diferentes estados de porosidad y densidad. Las celdas de la red cristalina están articuladas en las esquinas del tetrámero C98RhuA, permitiendo que gire y abra o cierre el poro. Crédito:Robert Alberstein et al.
¿Qué hace que kevlar detenga una bala? a nivel atómico?
Las propiedades de los materiales surgen de su estructura molecular o atómica, sin embargo, muchos detalles entre lo micro y lo macro siguen siendo un misterio para la ciencia. Los científicos están investigando activamente el diseño racional de arquitecturas supramoleculares específicas, con el objetivo de diseñar su dinámica estructural y su respuesta a las señales ambientales.
Un equipo de químicos de la Universidad de California, San Diego (UCSD) ha diseñado ahora un cristal de proteína bidimensional que alterna entre estados de porosidad y densidad variables. Esta es la primera vez en diseño biomolecular que combinó estudios experimentales con computación realizada en supercomputadoras. La investigación, publicado en abril de 2018 en Química de la naturaleza , podría ayudar a crear nuevos materiales para la energía renovable, medicamento, Purificación del agua, y más.
"Hicimos un amplio conjunto de simulaciones y experimentos de dinámica molecular, que explica la base de la inusual dinámica estructural de estas proteínas artificiales, en base a lo cual pudimos tomar decisiones racionales y alterar la dinámica estructural del ensamblaje, "dijo el coautor del estudio, Akif Tezcan, profesor de química y bioquímica en UCSD.
El equipo de Tezcan trabajó con la proteína L-ramnulosa-1-fosfato aldolasa (RhuA), que se modificó con aminoácidos de cisteína en sus cuatro esquinas en la posición 98 (C98RhuA). Él y su grupo habían publicado previamente un trabajo sobre el autoensamblaje de este artificial, arquitectura de proteínas bidimensional, que dijo mostraba un comportamiento interesante llamado auxeticidad.
"Estos conjuntos cristalinos pueden realmente abrirse y cerrarse en coherencia, "Tezcan dijo." Como lo hacen, se encogen o expanden por igual en las direcciones X e Y, que es lo contrario de lo que hacen los materiales normales. Queríamos investigar a qué se deben estos movimientos y qué los gobierna ". Un ejemplo de auxética se puede ver en la Esfera Hoberman, una pelota de juguete que se expande a través de sus bisagras en forma de tijera cuando separas los extremos.
"Nuestro objetivo era poder hacer lo mismo, utilizando proteínas como bloques de construcción, para crear nuevos tipos de materiales con propiedades avanzadas, ", Dijo Tezcan." El ejemplo que estamos estudiando aquí fue esencialmente el fruto de esos esfuerzos, donde usamos esta proteína en particular que tiene una forma cuadrada, que unimos unos a otros a través de enlaces químicos que eran reversibles y actuaban como bisagras. Esto permitió que estos materiales formaran cristales muy bien ordenados que también eran dinámicos debido a la flexibilidad de estos enlaces químicos. que acabó dándonos estas nuevas, propiedades emergentes."
El control de la apertura y cierre de los poros en las redes 2-D de la proteína C98RhuA podría capturar o liberar dianas moleculares específicas útiles para la administración de fármacos o la creación de mejores baterías con más investigación. Dijo Tezcan. O podrían atravesar o bloquear selectivamente el paso de moléculas biológicas y filtrar el agua.

La supercomputadora Maverick es un recurso dedicado de visualización y análisis de datos en el Centro de Computación Avanzada de Texas que está diseñado con 132 unidades de procesamiento de gráficos (GPU) NVIDIA Tesla K40 'Atlas' para visualización remota y computación GPU para la comunidad nacional. Crédito:TACC
"Nuestra idea era poder construir materiales complejos, como lo ha hecho la evolución, utilizando proteínas como bloques de construcción, "Dijo Tezcan.
La forma en que lo hizo el equipo de Tezcan fue expresar primero las proteínas en las células de la bacteria E. coli y purificarlas, después de lo cual indujeron la formación de enlaces químicos que realmente crean los cristales de C98RhuA, que varían en función de su estado de oxidación, mediante la adición de productos químicos con actividad redox.
"Una vez que se forman los cristales, la gran caracterización se convierte en la apertura o cercanía de los propios cristales, "explicó Tezcan, que se determinó mediante el análisis estadístico de cientos de imágenes capturadas mediante microscopía electrónica.
Los experimentos trabajaron de la mano con la computación, principalmente simulaciones de todos los átomos utilizando el software NAMD desarrollado en la Universidad de Illinois en Urbana Champaign por el grupo del difunto biofísico Klaus Schulten.
El equipo de Tezcan utilizó un sistema reducido de solo cuatro proteínas unidas entre sí, que se puede embaldosar infinitamente para llegar al fondo de cómo se abre y se cierra el cristal. "El sistema reducido nos permitió hacer estos cálculos factibles para nosotros, porque todavía hay cientos de miles de átomos, incluso en este sistema reducido, ", Dijo Tezcan. Su equipo aprovechó las funciones específicas de C98RhuA, como usar una sola coordenada de reacción correspondiente a su apertura. "Realmente pudimos validar este modelo como representativo de lo que observamos en el experimento, "Dijo Tezcan.
Las simulaciones moleculares de todos los átomos de las redes cristalinas C98RhuA se utilizaron para mapear el paisaje de energía libre. Este paisaje energético parece un paisaje natural, con valles, montañas, y pasos de montaña, explicó el coautor del estudio Francesco Paesani, profesor de química y bioquímica en UCSD.
"Los valles se convierten en las configuraciones más estables de sus conjuntos de proteínas, "Paesani dijo, que el sistema molecular prefiere a tener que gastar energía para cruzar una montaña. Y los pasos de montaña muestran el camino de una estructura estable a otra.
"Típicamente, Los cálculos de energía libre son muy costosos y desafiantes porque esencialmente lo que está tratando de hacer es muestrear todas las configuraciones posibles de un sistema molecular que contiene miles de átomos. Y desea saber cuántas posiciones pueden adquirir estos átomos durante una simulación. Se necesita mucho tiempo y muchos recursos informáticos, "Dijo Paesani.
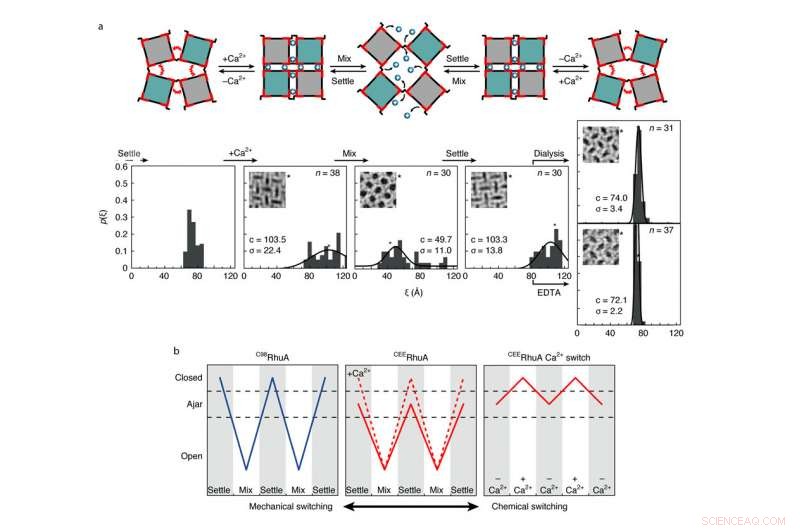
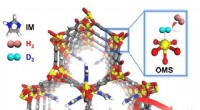
Comportamiento químico y mecánico de conmutación de cristales CEERhuA. a, Arriba:esquema que muestra todos los posibles modos de conmutación de las celosías CEERhuA. Abajo:distribución (es) experimental (es) correspondientes a los estados directamente arriba. La adición de 20 mM de Ca2 + a la población de cristales de CEERhuA 'entreabierta' en equilibrio indujo un cambio hacia conformaciones más cerradas, a partir del cual fue posible la conmutación mecánica similar a C98RhuA. La conformación entreabierta fue completamente recuperable al eliminar el Ca2 + mediante diálisis o EDTA, proporcionando así tres modos de conmutación distintos. Los ajustes gaussianos para cada distribución se etiquetan con su centro (c) y s.d. (σ). n es el número de cristales analizados. La conformación de celosía de cada inserción está marcada con un asterisco. B, Resumen de modos de conmutación para cristales RhuA. En contraste con C98RhuA, CEERhuA tiene dos modos mecánicos dictados por la presencia de Ca2 +, así como un modo puramente químico mediante la adición o eliminación de Ca2 +. Crédito:Robert Alberstein et al.
Para hacer frente a estos y otros desafíos computacionales, Paesani ha recibido asignaciones de supercomputadoras a través de XSEDE, el entorno de descubrimiento de ciencias e ingeniería extremas, financiado por la National Science Foundation.
"Afortunadamente, XSEDE nos ha proporcionado una asignación en Maverick, los clústeres de computación GPU en el Centro de Computación Avanzada de Texas (TACC), ", Dijo Paesani. Maverick es un recurso dedicado de visualización y análisis de datos diseñado con 132 unidades de procesamiento de gráficos (GPU) NVIDIA Tesla K40" Atlas "para visualización remota y computación GPU para la comunidad nacional.
"Eso fue muy útil para nosotros, porque el software NAMD que usamos funciona muy bien en las GPU. Eso nos permite acelerar los cálculos en órdenes de magnitudes, "Dijo Paesani." Hoy en día, podemos permitirnos cálculos con los que hace diez años ni siquiera podíamos soñar debido a estos desarrollos, tanto en el software NAMD como en el hardware. Todos estos grupos de computación que proporciona XSEDE son en realidad bastante útiles para todas las simulaciones dinámicas moleculares ".
A través de XSEDE, Paesani utilizó varios sistemas de supercomputación, incluido Gordon, Cometa, y Trestles en el San Diego Supercomputer Center; Kraken en el Instituto Nacional de Ciencias Computacionales; y Ranger, Estampida, y Stampede2 en TACC.
"Como todas las simulaciones se ejecutaron en GPU, Maverick fue la elección perfecta para este tipo de aplicación, "Dijo Paesani.
La computación y el experimento trabajaron juntos para producir resultados. "Creo que este es un hermoso ejemplo de la sinergia entre la teoría y el experimento, "Dijo Paesani." Experimento planteó la primera pregunta. La teoría y la simulación por computadora abordaron esa pregunta, proporcionando cierta comprensión del mecanismo. Y luego usamos simulación por computadora para hacer predicciones y pedir a los experimentos que probaran la validez de estas hipótesis. Todo salió muy bien porque las simulaciones explicaron los experimentos al principio. Las predicciones que se hicieron fueron confirmadas por los experimentos al final. Es un ejemplo de la sinergia perfecta entre experimentos y modelos teóricos ".
Tezcan agregó que "a los químicos tradicionalmente les gusta construir moléculas complejas a partir de bloques de construcción más simples, y uno puede imaginarse haciendo tal combinación de diseño, Experimento y cálculo de moléculas más pequeñas para predecir su comportamiento. Pero el hecho de que podamos hacerlo con moléculas compuestas por cientos de miles de átomos no tiene precedentes ".
El equipo científico también utilizó simulaciones de dinámica molecular para investigar rigurosamente el papel del agua en la dirección del movimiento reticular de C98RhuA. "Este estudio nos mostró lo importante que es el papel activo del agua en el control de la dinámica estructural de macromoléculas complejas, que en bioquímica puede pasarse por alto, ", Dijo Tezcan." Pero este estudio mostró, muy claro, que la dinámica de estas proteínas es impulsada activamente por la dinámica del agua, lo que creo que resalta la importancia del agua ".
Rob Alberstein, estudiante de posgrado en el grupo Tezcan y primer autor del artículo de Nature Chemistry, agregó:"En el corazón de esta investigación está comprender cómo las propiedades de los materiales surgen de la estructura molecular o atómica subyacente. Es muy difícil de describir. En este caso, realmente buscamos establecer esa conexión tan claramente como pudiéramos entenderla nosotros mismos y realmente mostrar no solo como desde el experimento, donde podemos observar el comportamiento a macroescala de estos materiales, pero luego, con el cálculo, relacione ese comportamiento con lo que realmente está sucediendo a escala de moléculas. A medida que continuamos desarrollándonos como sociedad, Necesitamos desarrollar nuevos materiales para nuevos tipos de problemas globales (purificación de agua, etc), por lo que comprender esta relación entre la estructura atómica y la propiedad material en sí y la capacidad de predecirlos será cada vez más importante ".