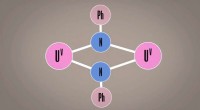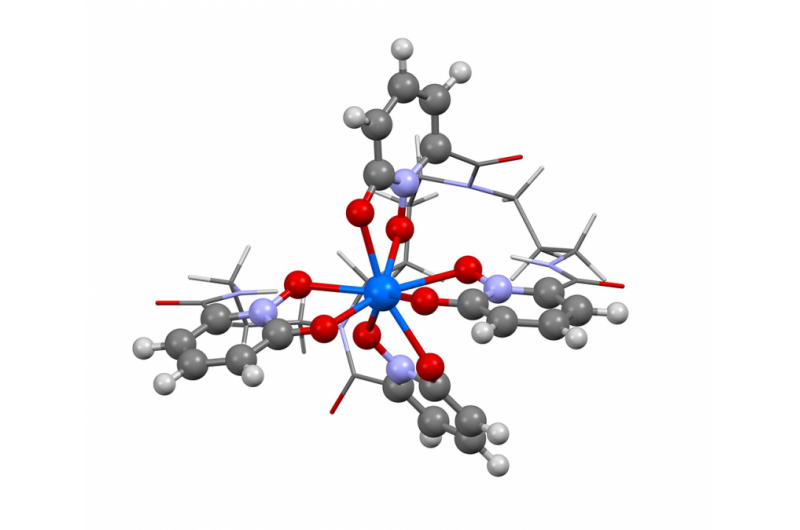
Esta imagen muestra la estructura del berkelio en estado de oxidación + IV. Los investigadores utilizaron el nuevo algoritmo de Berkeley Lab para calcular el espectro de absorción y confirmar lo que varios resultados experimentales han estado insinuando:que el elemento berkelio se rompe con sus pares del elemento pesado al adquirir una carga positiva adicional cuando se une a una molécula orgánica sintética. Esta propiedad podría ayudar a los científicos a desarrollar mejores métodos para manipular y purificar materiales nucleares. Crédito:Bert de Jong, Laboratorio de Berkeley
Los objetos que brillan en la oscuridad parecen mágicos cuando eres un niño:pueden iluminar una habitación oscura sin necesidad de electricidad, pilas o una bombilla. Luego, en algún momento, aprendes la ciencia detrás de este fenómeno. Los compuestos químicos llamados cromóforos se energizan, o emocionado, cuando absorben la luz visible. Cuando regresan a su estado normal, la energía almacenada se libera en forma de luz, que percibimos como un resplandor. En ciencia de materiales, Los investigadores se basan en un fenómeno similar para estudiar las estructuras de los materiales que eventualmente se utilizarán en la catálisis química. baterías aplicaciones solares y más.
Cuando una molécula absorbe un fotón, la partícula fundamental de luz, los electrones del sistema molecular pasan de un estado de baja energía (fundamental) a un estado de mayor energía (excitado). Estas respuestas resuenan en frecuencias de luz específicas, dejando "huellas digitales espectrales" que iluminan las estructuras atómicas y electrónicas del sistema en estudio.
En experimentos, las "huellas dactilares espectrales" o espectro de absorción, se miden con instalaciones de vanguardia como la fuente de luz avanzada (ALS) en el Laboratorio Nacional Lawrence Berkeley del Departamento de Energía de EE. UU. (Berkeley Lab). En simulaciones por computadora, estas medidas se capturan típicamente con un método mecánico cuántico llamado Teoría funcional de la densidad dependiente del tiempo (TDDFT). Los modelos computacionales son fundamentales para ayudar a los investigadores a aprovechar al máximo sus experimentos al predecir y validar resultados.
Sin embargo, a pesar de su utilidad, hay ocasiones en las que TDDFT no se puede utilizar para calcular el espectro de absorción de un sistema porque requeriría demasiado tiempo y recursos informáticos. Aquí es donde resulta útil un nuevo "atajo" matemático desarrollado por investigadores de la División de Investigación Computacional (CRD) de Berkeley Lab. Su algoritmo acelera los cálculos de absorción en un factor de cinco, de modo que las simulaciones que solían tardar entre 10 y 15 horas en calcularse ahora se pueden realizar en aproximadamente 2,5 horas.
Un artículo que describe este método se publicó en el Journal of Chemical Theory and Computation (JCTC). Y el nuevo enfoque para calcular el espectro de absorción se incorporará en una próxima versión del paquete de software de química computacional NWChem ampliamente utilizado a finales de este año.
Los nuevos algoritmos conducen a ahorros computacionales
Estudiar la estructura química de nuevas moléculas y materiales, los científicos normalmente sondean el sistema con un estímulo externo, normalmente un láser, y luego buscan pequeños cambios electrónicos. Matemáticamente, este cambio electrónico puede expresarse como un problema de valor propio. Al resolver este problema de valores propios, los investigadores pueden obtener una buena aproximación del espectro de absorción, que a su vez revela las frecuencias de resonancia del sistema en estudio. Mientras tanto, el vector propio correspondiente se utiliza para calcular con qué intensidad respondió el sistema al estímulo. Este es esencialmente el principio detrás del enfoque TDDFT, que se ha implementado en varios paquetes de software de química cuántica, incluido el paquete de software de código abierto NWChem.
Si bien este enfoque ha demostrado ser exitoso, tiene limitaciones para sistemas grandes. Cuanto más amplio sea el rango de energía de las respuestas electrónicas que un investigador intenta capturar en un sistema, Cuantos más autovalores y autovectores necesiten calcularse, lo que también significa que se necesitan más recursos informáticos. Por último, el espectro de absorción de un sistema molecular con más de 100 átomos se vuelve prohibitivamente caro de calcular con este método.
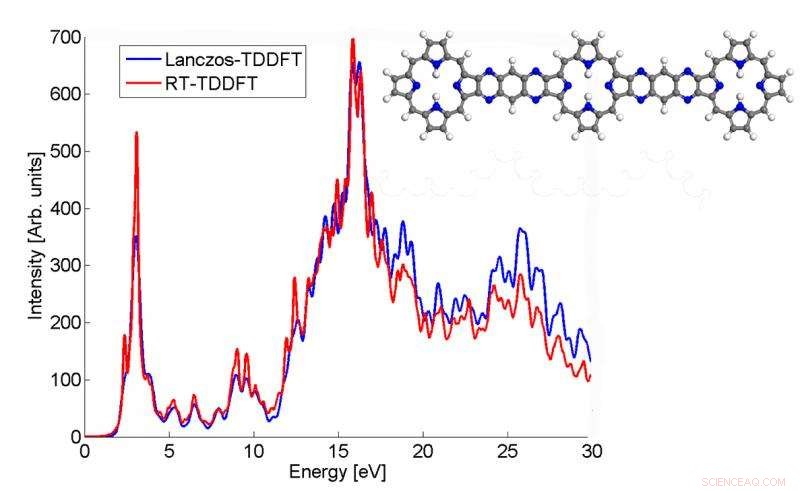
Este gráfico muestra cómo el espectro de absorción de una molécula p3b2 calculado por el algoritmo de Lanczos coincide con el resultado TDDFT en tiempo real. Crédito:Chao Yang, Laboratorio de Berkeley
Para superar estas limitaciones, Los matemáticos de CRD desarrollaron una técnica para calcular el espectro de absorción directamente sin calcular explícitamente los valores propios de la matriz.
"Tradicionalmente, los investigadores han tenido que calcular los valores propios y los vectores propios de matrices muy grandes para generar el espectro de absorción, pero nos dimos cuenta de que no es necesario calcular todos los valores propios para obtener una vista precisa del espectro de absorción, "dice Chao Yang, un matemático de CRD que dirigió el desarrollo del nuevo enfoque.
Al reformular el problema como una aproximación de función matricial, haciendo uso de una transformación especial y aprovechando la simetría subyacente con respecto a una métrica no euclidiana, Yang y sus colegas pudieron aplicar el algoritmo de Lanczos y un método polinomial Kernal (KPM) para aproximar el espectro de absorción de varias moléculas. Ambos algoritmos requieren una memoria relativamente baja en comparación con las alternativas no simétricas, que es la clave del ahorro computacional.
Debido a que este método requiere menos potencia informática para lograr un resultado, los investigadores también pueden calcular fácilmente el espectro de absorción para sistemas moleculares con varios cientos de átomos.
"Este método es un importante paso adelante porque nos permite modelar el espectro de absorción de sistemas moleculares de cientos de átomos a un menor costo computacional". dice Niranjan Govind, químico computacional del Pacific Northwest National Laboratory que colaboró con el equipo del Berkeley Lab en el desarrollo del método en el programa de química computacional NWChem.
Recientemente, los científicos de Berkeley Lab utilizaron este método para calcular el espectro de absorción y confirmar lo que varios resultados experimentales han estado insinuando:que el elemento berkelio se rompe con sus pares del elemento pesado al adquirir una carga positiva adicional cuando se une a una molécula orgánica sintética. Esta propiedad podría ayudar a los científicos a desarrollar mejores métodos para manipular y purificar materiales nucleares. Un artículo que destaca este resultado apareció el 10 de abril en la revista. Química de la naturaleza .
"Los resultados experimentales apuntaban a este comportamiento inusual en el berkelio, pero no hubo suficiente evidencia experimental para decir que sí, 100 por ciento, esto es lo que estamos viendo "dice el coautor del estudio, Wibe Albert de Jong, un científico de CRD. "Para estar 100% seguro, Hicimos grandes simulaciones computacionales y las comparamos con los datos experimentales y determinamos que eran, Por supuesto, ver berkelio en un estado de oxidación inusual ".
Este nuevo algoritmo fue desarrollado a través de un proyecto de Descubrimiento Científico a través de Computación Avanzada (SciDAC) respaldado por la Oficina de Ciencia del DOE, enfocado en el avance de software y algoritmos para reacciones fotoquímicas. Los proyectos SciDAC generalmente reúnen a un equipo interdisciplinario de investigadores para desarrollar métodos computacionales nuevos y novedosos para abordar algunos de los problemas científicos más desafiantes.
"La naturaleza interdisciplinaria de SciDAC es una forma muy eficaz de facilitar avances científicos, ya que cada miembro del equipo aporta una perspectiva diferente a la resolución de problemas, "dice Yang". En este entorno dinámico, matemáticos, como yo, formar equipo con científicos del dominio para identificar cuellos de botella computacionales, luego usamos técnicas matemáticas de vanguardia para abordar y superar esos desafíos ".