Un equipo de investigación del Instituto de Química Orgánica y Bioquímica de la Academia Checa de Ciencias/IOCB Praga ha desarrollado un nuevo método computacional que puede describir con precisión cómo las proteínas interactúan con las moléculas de fármacos potenciales y puede hacerlo en apenas decenas de minutos. Esta nueva función de puntuación de la mecánica cuántica puede acelerar considerablemente la búsqueda de nuevos fármacos. La investigación ha sido publicada en la revista Nature Communications. .
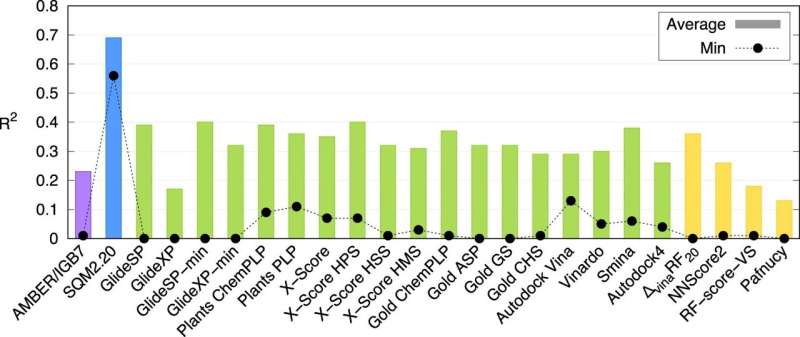
El estudio demuestra que este es el primer método de este tipo de aplicación universal. Los expertos computacionales del IOCB Praga lo probaron en 10 proteínas de diferentes niveles de complejidad estructural, cada una de las cuales une una gran variedad de moléculas pequeñas (generalmente denominadas ligandos). Luego compararon sus resultados no sólo con los de otros métodos correspondientes, sino también con los resultados de experimentos de laboratorio, y ambas comparaciones resultaron muy favorables.
"Por supuesto, no somos los únicos que trabajamos en esto. Existen varios métodos de este tipo. Sin embargo, normalmente su velocidad se ve compensada por una baja precisión, mientras que cálculos más precisos pueden tardar varios días. Nuestros métodos son únicos porque pueden procesar información sobre grandes sistemas moleculares en decenas de minutos, manteniendo al mismo tiempo las ventajas de cálculos mecánico-cuánticos mucho más exigentes", explica Jan Řezáč, autor correspondiente del artículo del grupo Interacciones no covalentes dirigido por el profesor Pavel Hobza.
Los expertos de este grupo llevan mucho tiempo estudiando las interacciones intermoleculares. En esta investigación se centran principalmente en biomoléculas y los resultados de su trabajo influyen directamente en el diseño de fármacos asistido por ordenador. La razón es que cuando los científicos trabajan en un nuevo fármaco, a menudo buscan moléculas que se unan fuertemente a una proteína en particular.
Sin embargo, identificarlas es como encontrar agujas en un pajar, ya que es necesario probar un gran número de moléculas para diferenciar aquellas que son prometedoras. Esto ralentiza considerablemente el descubrimiento de sustancias medicinales y lo encarece. Al predecir la fuerza de la unión proteína-ligando y, por tanto, seleccionar las moléculas que mejor satisfacen un conjunto definido de criterios, los químicos computacionales ahorran el trabajo de los experimentadores, lo que, a su vez, acelera significativamente el descubrimiento de fármacos.
Más información: Adam Pecina et al, SQM2.20:La función de puntuación mecánica cuántica semiempírica produce predicciones de afinidad de unión de ligando-proteína de calidad DFT en minutos, Nature Communications (2024). DOI:10.1038/s41467-024-45431-8
Información de la revista: Comunicaciones sobre la naturaleza
Proporcionado por Instituto de Química Orgánica y Bioquímica del CAS