Investigadores de la Clínica Cleveland e IBM han publicado recientemente hallazgos en el Journal of Chemical Theory and Computation. eso podría sentar las bases para aplicar métodos de computación cuántica a la predicción de la estructura de proteínas.
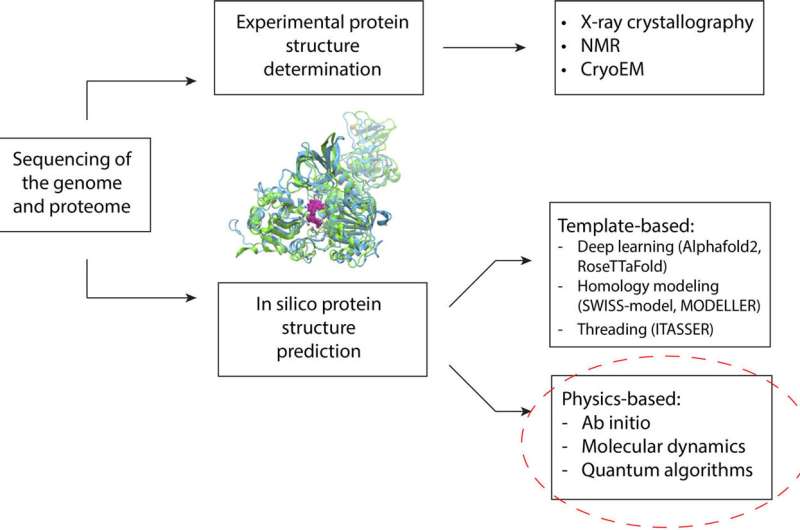
Durante décadas, los investigadores han aprovechado enfoques computacionales para predecir estructuras de proteínas. Una proteína se pliega en una estructura que determina cómo funciona y se une a otras moléculas del cuerpo. Estas estructuras determinan muchos aspectos de la salud y la enfermedad humanas.
Al predecir con precisión la estructura de una proteína, los investigadores pueden comprender mejor cómo se propagan las enfermedades y, por tanto, cómo desarrollar terapias eficaces. Bryan Raubenolt, Ph.D., becario postdoctoral de la Clínica Cleveland. y el investigador de IBM Hakan Doga, Ph.D. encabezó un equipo para descubrir cómo la computación cuántica puede mejorar los métodos actuales.
En los últimos años, las técnicas de aprendizaje automático han logrado avances significativos en la predicción de la estructura de las proteínas. Estos métodos dependen de datos de entrenamiento (una base de datos de estructuras de proteínas determinadas experimentalmente) para hacer predicciones. Esto significa que están limitados por la cantidad de proteínas que se les ha enseñado a reconocer. Esto puede llevar a niveles más bajos de precisión cuando los programas/algoritmos encuentran una proteína que está mutada o es muy diferente de aquella en la que fueron entrenados, lo cual es común en los trastornos genéticos.
El método alternativo consiste en simular la física del plegamiento de proteínas. Las simulaciones permiten a los investigadores observar las diversas formas posibles de una proteína determinada y encontrar la más estable. La forma más estable es fundamental para el diseño de fármacos.
El desafío es que estas simulaciones son casi imposibles en una computadora clásica, más allá de un cierto tamaño de proteína. En cierto modo, aumentar el tamaño de la proteína objetivo es comparable a aumentar las dimensiones de un cubo de Rubik. Para una proteína pequeña con 100 aminoácidos, una computadora clásica necesitaría un tiempo igual a la edad del universo para buscar exhaustivamente todos los resultados posibles, dice el Dr. Raubenolt.
Para ayudar a superar estas limitaciones, el equipo de investigación aplicó una combinación de métodos de computación clásica y cuántica. Este marco podría permitir que los algoritmos cuánticos aborden áreas que suponen un desafío para la informática clásica de última generación, incluido el tamaño de las proteínas, el desorden intrínseco, las mutaciones y la física implicada en el plegamiento de las proteínas. El marco se validó prediciendo con precisión el plegamiento de un pequeño fragmento de una proteína del virus Zika en una computadora cuántica, en comparación con los métodos clásicos de última generación.
Los resultados iniciales del marco híbrido cuántico-clásico superaron tanto a un método basado en la física clásica como a AlphaFold2. Aunque este último está diseñado para funcionar mejor con proteínas más grandes, demuestra la capacidad de este marco para crear modelos precisos sin depender directamente de datos de entrenamiento sustanciales.
Los investigadores utilizaron un algoritmo cuántico para modelar primero la conformación de menor energía para la columna vertebral del fragmento, que suele ser el paso del cálculo más exigente desde el punto de vista computacional. Luego se utilizaron enfoques clásicos para convertir los resultados obtenidos de la computadora cuántica, reconstruir la proteína con sus cadenas laterales y realizar el refinamiento final de la estructura con campos de fuerza de la mecánica molecular clásica.
El proyecto muestra una de las formas en que los problemas se pueden deconstruir en partes, con métodos de computación cuántica abordando algunas partes y computación clásica con otras, para lograr una mayor precisión.
"Una de las cosas más singulares de este proyecto es la cantidad de disciplinas involucradas", dice el Dr. Raubenolt. "La experiencia de nuestro equipo abarca desde biología y química computacional, biología estructural, software e ingeniería de automatización, hasta física atómica y nuclear experimental, matemáticas y, por supuesto, computación cuántica y diseño de algoritmos. Se necesitó el conocimiento de cada una de estas áreas para crear un marco computacional que puede imitar uno de los procesos más importantes para la vida humana."
La combinación del equipo de métodos de computación clásica y cuántica es un paso esencial para avanzar en nuestra comprensión de las estructuras de las proteínas y cómo afectan nuestra capacidad para tratar y prevenir enfermedades. El equipo planea continuar desarrollando y optimizando algoritmos cuánticos que puedan predecir la estructura de proteínas más grandes y sofisticadas.
"Este trabajo es un importante paso adelante en la exploración de dónde las capacidades de la computación cuántica podrían mostrar fortalezas en la predicción de la estructura de las proteínas", dice el Dr. Doga. "Nuestro objetivo es diseñar algoritmos cuánticos que puedan encontrar cómo predecir las estructuras de las proteínas de la manera más realista posible".
Más información: Hakan Doga et al, Una perspectiva sobre la predicción de la estructura de las proteínas utilizando computadoras cuánticas, Revista de teoría química y computación (2024). DOI:10.1021/acs.jctc.4c00067
Proporcionado por Cleveland Clinic