En un artículo publicado recientemente en Nature Communications , el grupo de investigación de modelado de proteínas HUN-REN-ELTE (Instituto de Química) ha sentado las bases de un método matemático que permite comparar, asistida por ordenador, las estructuras tridimensionales de las proteínas. El método es único en el sentido de que, mientras que las alternativas disponibles hasta ahora sólo tenían en cuenta la posición de los átomos, la nueva técnica, llamada LoCoHD (Distancia de Hellinger de composición local), también incluye la información química de los átomos.
Las proteínas son máquinas moleculares que llevan a cabo procesos necesarios para el funcionamiento de las células, actuando como interruptores moleculares, transcribiendo información del ADN, transportando moléculas pequeñas y grandes y regulando las reacciones químicas relacionadas con el metabolismo. Sin embargo, para que todo esto tenga éxito, la proteína en cuestión debe tener la conformación espacial correcta, es decir, su propia y correcta disposición 3D.
Se encuentran disponibles varios métodos experimentales (cristalografía de rayos X, espectroscopia de resonancia magnética nuclear, microscopía crioelectrónica) para determinar la disposición de los átomos en una proteína y, en las últimas décadas, los investigadores de proteínas han descubierto la forma de casi 220.000 proteínas. Estos resultados exigen cada vez más el desarrollo de métodos computacionales capaces de analizar estos arreglos.
Uno de esos métodos es el algoritmo llamado LoCoHD, desarrollado por Zsolt Fazekas, Ph.D. candidato de la Escuela de Química ELTE Hevesy György e investigador del grupo de investigación del Dr. András Perczel. El algoritmo compara los entornos locales alrededor de los aminoácidos en las proteínas en función de su naturaleza química (por ejemplo, composición elemental, carga, hidrofobicidad, etc.).
El método decide en una escala simple de 0 a 1 qué tan diferentes son las estructuras en cuestión entre sí. Los valores cercanos a 0 sugieren una gran similitud entre las disposiciones atómicas y las propiedades químicas, mientras que los valores cercanos a 1 indican que las proteínas que se comparan pueden tener propiedades muy diferentes. De este modo, el valor numérico resultante (lo que se conoce como métrica) se puede utilizar para obtener nueva información sobre el sistema en estudio.
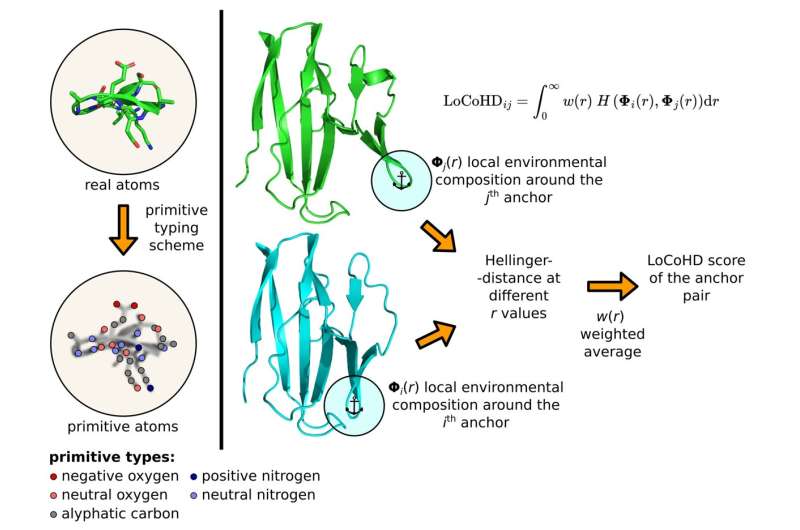
El algoritmo utiliza un protocolo de varios pasos para generar el número que representa las diferencias estructurales. En el primer paso, convierte los átomos reales de la proteína en los llamados átomos primitivos. Estos se pueden representar como posiciones virtualmente etiquetadas cuyas etiquetas indican la naturaleza química del átomo original.
Así, por ejemplo, un átomo primitivo puede ser un "nitrógeno con carga positiva", un "oxígeno con carga negativa", un "oxígeno con carga neutra", un "carbono aromático", etc. Las etiquetas se generan según el llamado átomo primitivo. esquema de tipificación, que nos dice de manera tabulada cómo convertir átomos reales en átomos primitivos. El usuario puede especificar libremente esta tabla, fijando la resolución química del método.
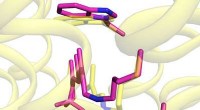
El segundo paso es determinar los puntos de referencia de la comparación seleccionando un subconjunto de átomos primitivos. Estos átomos primitivos especiales seleccionados se denominan átomos de anclaje. Para cada par de átomos de anclaje seleccionado, el algoritmo realiza un paso de comparación, cuyo resultado proporciona la medida de disimilitud que queremos. Estos números se pueden utilizar a nivel local o se pueden promediar en un único descriptor que caracterice la proteína completa.
En el estudio, los investigadores destacaron que el método también se puede utilizar en las competencias bianuales CASP (Evaluación crítica de la predicción de la estructura de las proteínas), que es una competencia muy conocida en el campo de la investigación de proteínas. Durante este evento, los competidores utilizan diferentes algoritmos para modelar la forma de proteínas que tienen estructuras aún inéditas. Los jueces del CASP utilizan varios métodos de comparación de estructuras para evaluar a los contendientes, pero ninguno de ellos tiene en cuenta la química de los entornos locales de aminoácidos.
Utilizando datos de la competencia CASP14 de 2020, los investigadores han realizado análisis comparativos de varias proteínas modeladas, incluidas las estructuras predichas por el método AlphaFold2 basado en inteligencia artificial. Entre estos destacaron el análisis de una proteína del virus SARS-CoV-2 llamada ORF8. En las estructuras modeladas de esta proteína, se identificaron entornos de aminoácidos que difieren significativamente en sus patrones de interacción de los entornos encontrados en la estructura experimental.
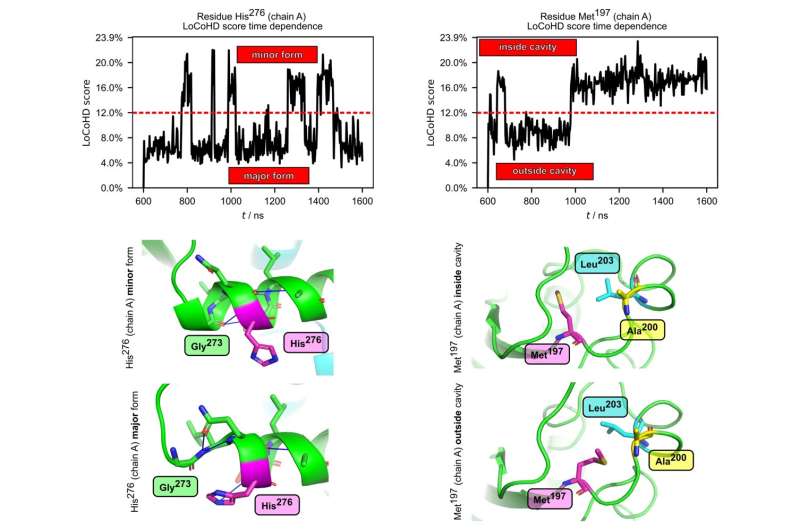
Además de estudiar las estructuras estáticas, los investigadores también comprobaron si el método es adecuado para analizar el movimiento interno de las proteínas. Utilizaron simulaciones capaces de reproducir movimientos moleculares y datos extraídos de conjuntos estructurales. Uno de los sistemas estudiados fue la proteína podocina, que realiza funciones vitales en el riñón y cuyas mutaciones pueden provocar enfermedades graves, a menudo mortales.
El método LoCoHD se utilizó para identificar los aminoácidos de la proteína que sufren importantes cambios químico-ambientales durante el movimiento de la podocina, que pueden afectar tanto a su estructura como a su función. Asimismo, el método LoCoHD se ha aplicado con éxito en el estudio de la proteína de la cápside del VIH-1, en la que se ha identificado un aminoácido crítico para la formación de la envoltura viral.
Estos resultados no son sólo curiosidades de la investigación, sino que al estudiar las estructuras de las proteínas de manera más efectiva, podemos acercarnos a una mejor comprensión de los patógenos que causan enfermedades graves y al desarrollo de fármacos y terapias eficaces.
Más información: Zsolt Fazekas et al, LoCoHD:una métrica para comparar entornos locales de proteínas, Nature Communications (2024). DOI:10.1038/s41467-024-48225-0
Información de la revista: Comunicaciones sobre la naturaleza
Proporcionado por la Universidad Eötvös Loránd