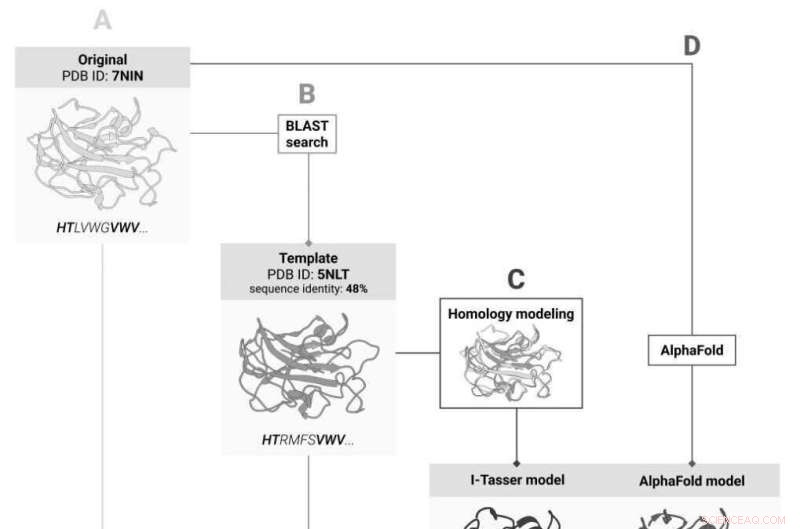
Cuatro formas de predecir cambios en la estabilidad de la proteína después de la mutación:(A) por la estructura de la proteína original; (B) por la estructura de su homólogo; (C) por la estructura de la proteína original predicha en base a la estructura del holomlog, y (D) por la estructura predicha por la inteligencia artificial en base a la secuencia de aminoácidos. Crédito:Instituto Skolkovo de Ciencia y Tecnología
Investigadores del Centro Skoltech de Biología Molecular y Celular compararon diferentes métodos de predicción de estructuras de proteínas en términos de evaluación de la estabilidad de proteínas mutantes y obtuvieron el mismo resultado para las estructuras predichas por IA y tridimensionales (3D) experimentales de proteínas con secuencias de aminoácidos similares. Sin embargo, el intento de predecir la estructura de la proteína objetivo a partir de la estructura conocida de su "pariente" solo hizo que la predicción fuera menos precisa. Los hallazgos del equipo facilitarán los cálculos preliminares en la evaluación de los cambios de estabilidad causados por la mutación. La investigación fue publicada en Bioinformatics .
Los experimentos biológicos a menudo involucran proteínas mutantes, que son necesarias para el estudio de la estructura y las funciones de las proteínas o los procesos celulares, así como para la ingeniería de proteínas. Se sabe que las mutaciones afectan la estructura y la estabilidad de una proteína. Dado que los experimentos son demasiado costosos y consumen mucho tiempo, los científicos están creando una solución en forma de métodos computacionales para evaluar el impacto de las mutaciones en la estabilidad. Sin embargo, sus aplicaciones requieren el conocimiento de la estructura 3D de una proteína.
Una estructura 3D experimental no está disponible para todas las proteínas y es probable que falte para la que el equipo tiene como objetivo. Si este es el caso, los modelos 3D de los homólogos de la proteína, es decir, sus "parientes más cercanos", pueden proporcionar el salvavidas, porque el grado de similitud en las secuencias de aminoácidos que asegura una buena coincidencia entre las estructuras 3D de las proteínas es bien conocido. La solución sería predecir primero la estructura de la proteína en función de la estructura conocida de su homólogo y luego calcular el impacto de las mutaciones para el modelo predicho.
Gracias al avance del año pasado en la predicción de la estructura de proteínas, los científicos ahora tienen una alternativa:en lugar de predecir la estructura 3D basándose en homólogos, pueden usar la herramienta AlphaFold basada en IA que predice la estructura de la proteína a partir de la secuencia de aminoácidos y ya ha tratado con la gran mayoría de proteínas conocidas hasta la fecha.
En su estudio reciente, los investigadores de Skoltech decidieron averiguar cuál de estos enfoques funciona mejor para predecir los cambios de estabilidad tras la mutación. Por muy preciso que sea AlphaFold, encontrar la estructura de la proteína a través de experimentos sigue siendo el "estándar de oro". Al comparar los dos enfoques, el equipo utilizó siete métodos de evaluación de la estabilidad y comparó sus resultados con los de AlphaFold e I-Tasser, el mejor sistema de predicción de estructuras basado en homólogos. Además, los investigadores comprobaron si pueden omitir la predicción de la estructura basada en homólogos y calcular la estabilidad de la estructura conocida de la proteína homóloga.
"Decidimos averiguar hasta qué punto nos desviaríamos de la predicción precisa si usáramos la estructura de la proteína 'vecina' en lugar de la real. Resultó que el paso de predicción basado en la homología solo empeora las cosas al producir un resultado menos preciso. Hemos demostrado que prácticamente no hay diferencia entre usar la estructura experimental del homólogo o la predicción de AlphaFold. En cierto sentido, se trataba de validación:cuando se enfrenta a un nuevo método, debe verificar si funciona para su tarea en primer lugar. Eso es exactamente lo que hicimos", dijo el primer autor del estudio, Skoltech Ph.D. estudiante Marina Pak, comentarios.
"Con todo este alboroto por AlphaFold, algunos científicos y no profesionales creen que ha resuelto todos los problemas de investigación de proteínas en biología computacional, pero no lo ha hecho. Por ejemplo, la predicción de los cambios de estabilidad inducidos por mutaciones todavía muestra una confiabilidad bastante baja, incluso aunque el cambio en la estabilidad es uno de los impulsores clave de la funcionalidad de la proteína. Una herramienta que pudiera determinar sin ambigüedades el impacto de la mutación en la estabilidad ayudaría tanto en la planificación del experimento como en la interpretación de los resultados. Supongamos que para una proteína que no es óptima en términos de estabilidad, deseamos encontrar mutaciones que lo hagan estable en las condiciones deseadas, por ejemplo, garantizar que permanezca activo a alta temperatura. Una vez que podamos hacer esto solo mediante cálculos, el enfoque para el rediseño y la optimización de proteínas cambiará drásticamente". autor principal del estudio, concluye el profesor asistente de Skoltech Dmitry Ivankov.
Aunque la predicción del cambio de estabilidad parece más fácil que la predicción de la estructura 3D, sigue siendo un desafío insuperable incluso para la IA. Los escasos datos de entrenamiento son solo uno de los problemas:AlphaFold tenía casi 200.000 estructuras de proteínas para entrenar, pero los datos experimentales sobre los cambios de estabilidad ascienden a miles de conjuntos y cubren solo unas pocas docenas de proteínas únicas. Los autores esperan que si hay más datos disponibles y los investigadores muestran un mayor interés en la tarea, es seguro que pronto se producirá un gran avance. Los físicos usan IA para encontrar los nudos de proteínas más complejos hasta el momento