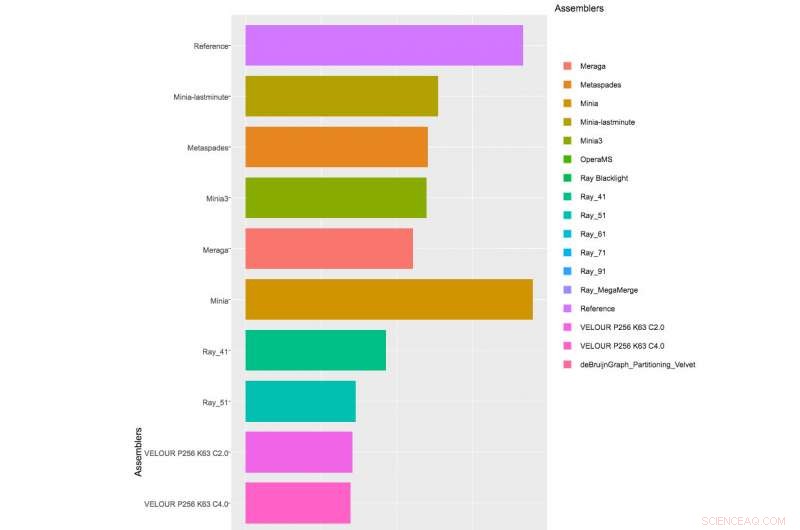
Tabla que muestra resultados parciales de ensambladores aplicados al 1er Desafío CAMI, Conjunto de datos 1. Haga clic aquí para ver la tabla completa.
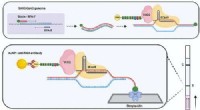
Están en todos lados, pero invisible a simple vista. Los microbios son lo invisible fuerzas influyentes detrás de la regulación de procesos ambientales clave como el ciclo del carbono, sin embargo, la mayoría de ellos siguen siendo desconocidos. Durante más de una década, el Instituto Conjunto del Genoma del Departamento de Energía de EE. UU. (DOE JGI), una instalación para usuarios de la Oficina de Ciencias del DOE, ha permitido a los investigadores estudiar microbios no cultivados que no pueden crecer en el laboratorio, utilizando enfoques de vanguardia como la secuenciación genómica de alto rendimiento de comunidades ambientales ("metagenómica") y el desarrollo de herramientas computacionales para descubrir y caracterizar comunidades microbianas del medio ambiente. Para abordar el ensamblaje de metagenomas en un conjunto de segmentos de ADN superpuestos que juntos representan una región de consenso de ADN o contigs, luego agrupando estos contigs en contenedores de genoma, y finalmente la realización de perfiles taxonómicos de los contenedores del genoma, analistas de todo el mundo han desarrollado una serie de diferentes herramientas computacionales, sin embargo, hasta ahora ha habido una falta de consenso sobre cómo evaluar su desempeño.
Publicado el 2 de octubre de 2017 en Métodos de la naturaleza , un equipo que incluye a investigadores del DOE JGI describió los resultados del Desafío de la Evaluación Crítica de la Interpretación del Metagenoma (CAMI), el primero en la historia evaluación comparativa organizada por la comunidad de herramientas computacionales para metagenomas. El CAMI Challenge fue dirigido por Alexander Sczyrba, jefe del grupo de Metagenómica Computacional en la Universidad de Bielefeld y anteriormente becario postdoctoral DOE JGI, y Alice McHardy, jefe del Laboratorio de Investigación de Biología Computacional de Infecciones en el Centro Helmholtz de Investigación de Infecciones.
"Es muy difícil para los investigadores averiguar qué programa utilizar para un conjunto de datos y un análisis en particular basándose en los resultados de los artículos del método, ", dijo McHardy." Los conjuntos de datos y las medidas de evaluación utilizadas en las evaluaciones varían ampliamente. Otro problema es que los desarrolladores suelen dedicar mucho tiempo a comparar el estado de la técnica cuando evalúan el rendimiento de un software novedoso de esa manera. CAMI quiere cambiar estas cosas e involucra a la comunidad en la definición de estándares y mejores prácticas para la evaluación y en la aplicación de estos principios en los desafíos de evaluación comparativa ".
El CAMI Challenge se llevó a cabo durante tres meses en 2015. Para evaluar las herramientas computacionales, Los organizadores desarrollaron 3 conjuntos de datos de metagenomas simulados utilizando más de 300 borradores de genomas de aislados de bacterias y arqueas secuenciados y ensamblados por el DOE JGI. que formaban parte del proyecto Genomic Encyclopedia of Bacteria and Archaeal publicado recientemente en Biotecnología de la naturaleza . Estos genomas se compartieron con el consorcio CAMI Challenges antes de ser lanzados al público para facilitar la evaluación comparativa objetiva de diferentes herramientas. Los conjuntos de datos también incluyeron aproximadamente la misma cantidad de genomas del Instituto Max Planck en Colonia, Alemania, junto con elementos circulares y virus. Los conjuntos de datos simulados fueron un conjunto de datos de muestra única de 15 mil millones de bases (Gb), un conjunto de datos de 40 Gb con 40 genomas y 20 elementos circulares, y un conjunto de datos de series de tiempo de 75 Gb compuesto por cinco muestras y que incluye cientos de genomas y elementos circulares.
"JGI tiene un gran interés en la evaluación comparativa de herramientas y tecnologías que avanzarían en el análisis de metagenomas y mejorarían la calidad de los datos que proporcionamos a los usuarios. Habiendo publicado el primer estudio sobre el uso de conjuntos de datos simulados para la evaluación comparativa de herramientas de metagenómica de JGI, Es genial ver cómo esta metodología se ha expandido a lo largo de los años y ahora a través de este estudio, evolucionando hacia un modelo para los esfuerzos comunitarios estandarizados en el campo, "dijo Nikos Kyrpides, Jefe del Super Programa Prokaryote DOE JGI.
"JGI está muy comprometido no solo con la evaluación comparativa de los protocolos de laboratorio, sino también flujos de trabajo computacionales, ", agregó la directora de Genómica Microbiana del DOE JGI, Tanja Woyke. Esto hace que nuestra participación en esfuerzos comunitarios críticos como CAMI sea tan importante".
Con más de 40 equipos inscritos en el Desafío, y los organizadores de CAMI recibieron 215 presentaciones de 25 programas en todo el mundo, aunque solo 17 equipos estaban dispuestos a publicar sus implementaciones de software. Los organizadores de CAMI evaluaron las herramientas computacionales en 3 categorías. Se evaluaron media docena de ensambladores y tuberías de ensamblaje en el ensamblaje de secuencias del genoma generadas a partir de tecnologías de secuenciación de lectura corta. En el desafío del binning, five genome binners and 4 taxonomic binners were evaluated on criteria including the tools' efficacy in recovering individual genomes. Finalmente, 10 taxonomic profilers with various parameter settings were evaluated on how well they could predict the identities and relative abundances of the microbes and circular elements. The benchmarking results are available on https://data.cami-challenge.org/results.
The CAMI organizers are already planning future benchmarking challenges, for example to evaluate and aid method development for long read sequencing technologies. "CAMI is an ongoing initiative, " noted Sczyrba. "We are currently further automating the benchmarking and comparative result visualizations. And we invite everyone interested to join and work with CAMI on providing comprehensive performance overviews of the computational metagenomics toolkit, to inform developers about current challenges in computational metagenomics and applied scientists of the most suitable software for their research questions."