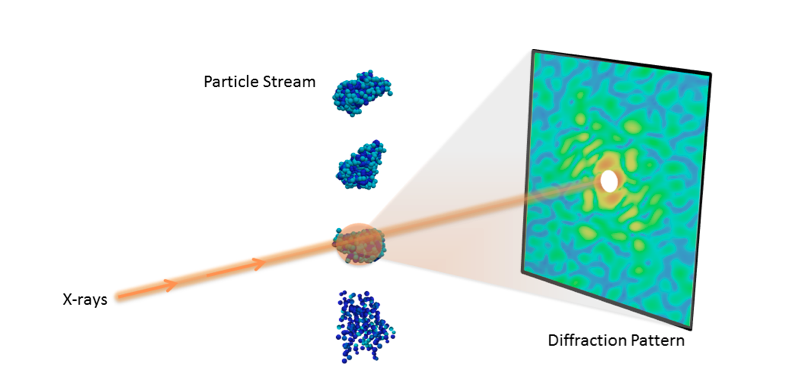
Configuración experimental para un experimento de difracción de una sola partícula. Crédito:Peter Zwart, Laboratorio de Berkeley
Comprender la estructura molecular 3D de nanoobjetos importantes como proteínas y virus es crucial en biología y medicina. Con los avances recientes en la tecnología de rayos X, los científicos ahora pueden recolectar imágenes de difracción de partículas individuales, permitiendo en última instancia a los investigadores visualizar moléculas a temperatura ambiente.
Sin embargo, determinar la estructura 3D a partir de estos experimentos de difracción de una sola partícula es un obstáculo importante. Por ejemplo, las tasas de adquisición de datos actuales son muy limitantes, normalmente resulta en menos de 10 instantáneas útiles por minuto, limitando la cantidad de características que se pueden resolver. Adicionalmente, las imágenes a menudo están muy corrompidas por ruido y otros artefactos experimentales, lo que dificulta la interpretación adecuada de los datos.
Para hacer frente a estos desafíos, un equipo de investigadores del Laboratorio Nacional Lawrence Berkeley (Berkeley Lab) ha desarrollado un nuevo marco algorítmico llamado fase iterativa de múltiples niveles (M-TIP) que utiliza técnicas matemáticas avanzadas para determinar la estructura molecular 3D a partir de conjuntos muy escasos de ruidos, datos de una sola partícula. Este enfoque esencialmente permite a los investigadores extraer más información de experimentos con datos limitados. Los matemáticos aplicados Jeffrey Donatelli y James Sethian, y el biocientífico físico Peter Zwart introdujeron este marco ampliando un algoritmo que desarrollaron originalmente para resolver la reconstrucción de un experimento de dispersión de rayos X relacionado, llamada dispersión de rayos X por fluctuación. Un artículo que describe el marco M-TIP se publicó el 26 de junio en el procedimientos de la Academia Nacional de Ciencias .
"Este enfoque tiene el potencial de revolucionar el campo, "dice Zwart." Dado que es difícil obtener una gran cantidad de buenos datos, Es probable que los enfoques que reducen la cantidad de datos necesarios para obtener imágenes de nanoobjetos 3D con éxito reciban una cálida bienvenida ".
Donatelli, Sethian y Zwart son parte de CAMERA (el Centro de Matemáticas Avanzadas para Aplicaciones de Investigación Energética), cuya misión es crear las matemáticas de vanguardia necesarias para manejar datos de muchas de las instalaciones científicas más avanzadas del DOE. CAMERA está financiada conjuntamente por los programas de Investigación en Computación Científica Avanzada y Ciencias de la Energía Básica en la Oficina de Ciencias del DOE.
Difracción de partículas individuales
El reciente advenimiento de los láseres de rayos X de electrones libres (XFEL) ha permitido varias técnicas experimentales nuevas para estudiar biomoléculas que no eran factibles con fuentes de luz tradicionales. Una de esas técnicas es la difracción de una sola partícula, que recopila una gran cantidad de instantáneas de difracción de rayos X con una sola partícula en el haz. Aprovechando la potencia extrema de los XFEL, los investigadores pueden recopilar señales mensurables incluso de las partículas más pequeñas.
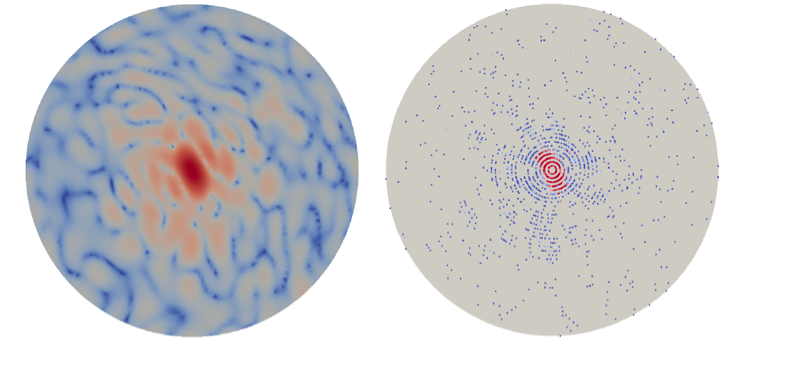
Un ejemplo de una imagen de difracción de una sola partícula limpia (izquierda) y la misma imagen de difracción después de la contaminación por ruido (derecha). Crédito:Peter Zwart, Laboratorio de Berkeley
Una gran ventaja que ofrece esta técnica de difracción de una sola partícula es la capacidad de estudiar cómo las diferentes copias de una molécula varían o cambian de forma. Dado que cada imagen proviene de una sola partícula, estas variaciones se pueden capturar en el experimento, a diferencia de los métodos tradicionales de obtención de imágenes como la cristalografía o la dispersión de rayos X de ángulo pequeño, donde los investigadores solo pueden medir un promedio en todos los diferentes estados de la muestra molecular.
Sin embargo, determinar la estructura 3D a partir de datos de difracción de una sola partícula es un desafío. Empezar, cuando se obtiene la imagen de cada partícula, su orientación es desconocida y debe recuperarse para combinar correctamente los datos en un volumen de difracción 3D. Este problema se agrava si la molécula puede adoptar diferentes formas, lo que requiere una clasificación adicional de las imágenes. Es más, La información de fase no se registra en imágenes de difracción y debe recuperarse para completar la reconstrucción. Finalmente, incluso con potentes XFEL, el número de fotones dispersos es muy pequeño, resultando en imágenes extremadamente ruidosas, que pueden contaminarse aún más por problemas sistemáticos de lectura de fondo y del detector.
Los enfoques anteriores se basan en resolver el problema de reconstrucción en pasos separados, donde cada problema individual se aborda por separado. Desafortunadamente, un inconveniente de estos enfoques en serie es que no aprovechan fácilmente las características conocidas anteriores sobre el aspecto de la molécula. Además, cualquier error cometido en un paso se propaga al siguiente, resultando en un aumento adicional del error. Esta "bola de nieve de error" finalmente degrada la calidad de la reconstrucción obtenida en el paso final.
Lo mejor de ambos mundos
En lugar de resolver los problemas de cálculo en pasos separados, El algoritmo M-TIP del equipo resuelve todas las partes del problema al mismo tiempo. Este enfoque aprovecha la información previa sobre la estructura para reducir en gran medida los grados de libertad del problema en todos los pasos, y consecuentemente reducir la información requerida necesaria para lograr una reconstrucción 3D.
"Las técnicas estándar de optimización de caja negra pueden incorporar conocimientos previos en la reconstrucción, pero descartan toda la estructura del problema, mientras que resolverlo en subpasos en serie completamente separados explota la estructura del problema pero descarta casi toda la información anterior sobre cómo podría ser la solución, ", Dijo Donatelli." M-TIP aprovecha lo mejor de ambos mundos explotando la estructura del problema para dividir el cálculo en varios fragmentos manejables y luego refinando iterativamente todos estos fragmentos para llegar a una solución que sea consistente con los datos y cualquier restricción estructural ".
Usando esta técnica, el equipo pudo determinar la estructura 3D a partir de recuentos de imágenes extremadamente bajos a partir de datos simulados, tan bajo como 6 a 24 imágenes para datos sin ruido y 192 imágenes de datos altamente contaminados.
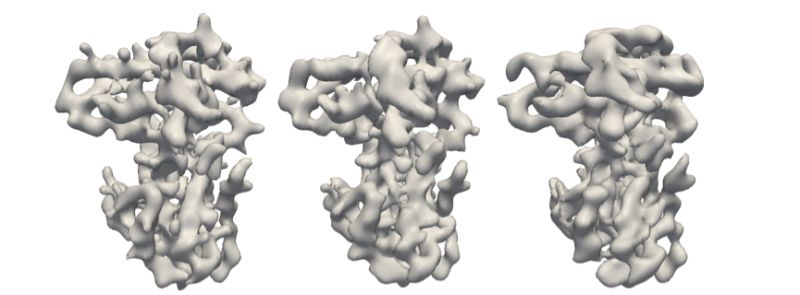
Proteína de retinoblastoma original (izquierda) y reconstrucciones utilizando el algoritmo M-TIP con 24 imágenes limpias (centro) y 192 imágenes ruidosas (derecha), como se muestra en la Figura 2. Crédito:Peter Zwart, Laboratorio de Berkeley
Abriendo nuevos caminos
Este trabajo es parte de una nueva iniciativa de colaboración entre SLAC National Accelerator Laboratory, CÁMARA, el Centro Nacional de Computación Científica de Investigación de Energía (NERSC) y el Laboratorio Nacional de Los Alamos como parte del Proyecto de Computación Exascale (ECP) del DOE. El objetivo del proyecto es desarrollar las herramientas computacionales necesarias para realizar análisis de datos en tiempo real a partir de experimentos que se llevan a cabo en la fuente de luz coherente Linac (LCLS) de SLAC. Con actualizaciones a la línea de luz, LCLS-II planea generar varios terabytes de datos por segundo, cuales, por ejemplo, permitirá a los científicos ampliar en gran medida los experimentos actuales de una sola partícula. El análisis de todos estos datos en tiempo real requerirá nuevos algoritmos y grandes máquinas informáticas. El algoritmo M-TIP servirá como parte de este proceso.
"Estos son algunos de los problemas más desafiantes en la ciencia de datos computacionales, "dice Sethian." Para abordarlos, necesitamos aprovechar una variedad de tecnologías, incluidas las arquitecturas informáticas emergentes de exaescala, sofisticadas redes de alta velocidad, y los algoritmos matemáticos más avanzados disponibles. Reunir a los científicos de CAMERA con proyectos de aplicación a exaescala ha abierto la puerta a la creación de herramientas para abordar algunos problemas urgentes en biología y ciencias de los materiales ".
Los investigadores señalan que estos son solo los primeros pasos. Para que el método esté listo para implementarse, hay que superar otros obstáculos.
"La ciencia experimental es complicada, ", dice Zwart." Hay efectos experimentales adicionales que deben tenerse en cuenta para que podamos obtener los mejores resultados posibles ".
"Afortunadamente, M-TIP es una técnica muy modular, "añade Donatelli, "asi que, es adecuado para modelar muchos de estos efectos adicionales sin necesidad de cambiar el marco algorítmico central ".
El equipo está trabajando actualmente en el estudio de estos efectos como parte de la iniciativa de partículas únicas, un gran, colaboración multiinstitucional dedicada a abordar problemas teóricos y prácticos en la obtención de imágenes de una sola molécula basada en X-FEL, lo que en última instancia conduce a proporcionar a la comunidad científica las herramientas necesarias para abrir nuevos caminos en biología, ciencias de la medicina y la energía.