
Pepsi-SAXS:nuevo método de análisis de proteínas que es 50 veces más rápido que los análogos. Crédito:MIPT
Pepsi-SAXS es un nuevo método altamente eficiente para el cálculo de perfiles de dispersión de rayos X, que son necesarios para el análisis de moléculas de proteínas en estado de solución. El método fue creado por científicos de la Université Grenoble Alpes y MIPT, dirigido por Sergei Grudinin. El equipo probó su método, y los resultados fueron publicados por la Unión Internacional de Cristalografía en su revista Acta Crystallographica Sección D:Biología estructural .
Las proteínas tienen una estructura compleja y un tamaño extremadamente pequeño, del orden de varios nanómetros. Para estudiarlos, los investigadores deben idear métodos inusuales, porque las muestras de proteínas se destruyen con demasiada facilidad y sus propiedades se alteran en los experimentos. El conocimiento de las estructuras y los mecanismos funcionales de las biomoléculas permite que se desarrollen nuevos fármacos, no por ensayo y error, lo que técnicamente se denomina cribado de alto rendimiento, sino de una manera más centrada.
Una de las técnicas que se utilizan para estudiar las proteínas es el análisis de los rayos X que se desprenden de ellas. Los investigadores necesitan usar rayos X y no luz ordinaria para acercar átomos individuales con un tamaño característico del orden de 0,1 nanómetros. Cuanto más pequeño es el objeto, cuanto más corta sea la longitud de onda de la luz que se debe utilizar para observarlo. La luz visible comprende longitudes de onda entre 400 y 700 nanómetros. Rayos X, por otra parte, tienen una longitud de onda mucho más corta y, por lo tanto, se pueden utilizar para examinar estructuras moleculares.
"El nuevo método nos permite trazar curvas de dispersión de manera eficiente y precisa, y analizar la estructura tridimensional de una muestra, "dice la estudiante de MIPT Maria Garkavenko, coautor del artículo. "Entre otras cosas, Pepsi-SAXS aumenta la eficiencia del modelado y la precisión de la predicción de la estructura de macromoléculas tridimensionales ".
Dispersión de rayos X de ángulo pequeño, o SAXS, es una técnica experimental que consiste en dispersar rayos X de una muestra y luego recolectarlos en ángulos muy pequeños. Como resultado, Se obtiene un gráfico de la intensidad del haz de rayos X disperso en función del ángulo de incidencia. Usando esta trama, una muestra de proteína se puede comparar con otras muestras en la base de datos experimental para determinar su estructura y propiedades.
En comparación con otras técnicas utilizadas para determinar la estructura de la muestra, SAXS es mucho más sencillo y económico. Requiere solo una preparación mínima de la muestra, y las proteínas no necesitan congelarse ni cristalizarse. Las muestras se estudian en solución y en su estado funcional. Esto hace que los resultados sean mucho más fiables, porque la preparación de la muestra a veces puede alterar el estado y las propiedades de una proteína. Otra ventaja importante del método es que no es destructivo, lo que significa que la muestra experimental no se ve afectada en gran medida por los rayos X.
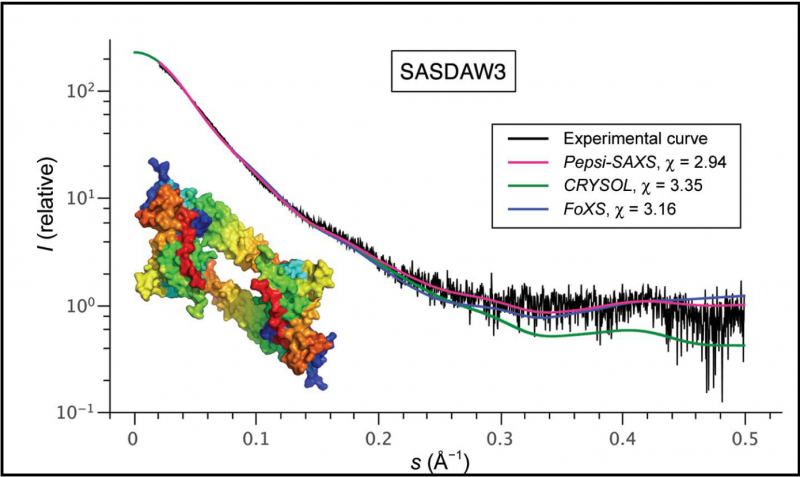
La Figura 1 muestra los resultados de una serie de experimentos, que comparó Pepsi-SAXS con dos de los métodos computacionales utilizados actualmente aplicándolos a la misma muestra (SASDAW3) de la base de datos SASBDB. La intensidad de dispersión media se representa en función del ángulo de dispersión. El error χ² del modelo computacional es el más bajo en el caso de Pepsi-SAXS, que resulta de una representación más precisa de la capa de hidratación. Crédito:S. Grudinin, M. Garkavenko y A. Kazennov
Pero hasta hace poco SAXS tenía un gran inconveniente:el método era computacionalmente intensivo, lo que significaba que no se podía utilizar si el número de experimentos era considerable. Se necesitaron horas para procesar los resultados de un solo experimento. Inicialmente, el número de cálculos fue directamente proporcional al cuadrado del número de átomos en la muestra, este último número suele superar el millar. Sin embargo, en los 1970s, Heinrich Stuhrmann, un investigador alemán, se le ocurrió una idea que simplificó los cálculos. Propuso que la dispersión en compuestos moleculares se describiera en términos de funciones de un tipo particular llamadas armónicos esféricos. Este enfoque resultó ser un éxito. A través de los años, Se crearon una serie de herramientas computacionales para el análisis de datos SAXS. Investigadores con antecedentes científicos soviéticos, entre ellos Dmitri Svergun (que actualmente trabaja en Hamburgo), hicieron contribuciones importantes a su desarrollo, quien escribió el paquete de software ATSAS para el análisis de datos SAXS en la investigación de macromoléculas biológicas. Los investigadores del estudio que se informa aquí examinaron varios métodos computacionales y los compararon con su propia técnica.
“Pepsi-SAXS significa 'expansiones polinomiales de estructuras e interacciones de proteínas' y 'dispersión de rayos X de ángulo pequeño'. Es un método adaptativo para el cálculo rápido y preciso de perfiles de dispersión de rayos X de ángulo pequeño, "explica el estudiante de doctorado del MIPT Andrei Kazennov, coautor del artículo. "Pepsi-SAXS se puede adaptar al tamaño de una muestra dada y la resolución de los datos experimentales".
Los investigadores también crearon un modelo eficiente de la capa de hidratación, una capa de moléculas de agua que rodean a las proteínas en solución, y lo incorporaron a su software. aumentando la precisión del método.
"Nuestro método ha sido validado en un gran conjunto de datos de BioIsis y SASBDB, las dos bases de datos biológicas más grandes, "dice Sergei Grudinin, quien supervisó la investigación. "Hemos demostrado que Pepsi-SAXS es de cinco a 50 veces más rápido que los métodos utilizados anteriormente, a saber, CRYSOL, FoXS, y la técnica de Zernike tridimensional implementada en el paquete SAStbx. Al mismo tiempo, la precisión está a la par con ellos ".
Los investigadores prestaron especial atención al análisis de los resultados obtenidos, que se compararon con los datos experimentales.
La investigación de proteínas tiene un significado fundamental para nuestra comprensión de los procesos básicos que subyacen a la vida, así como para el desarrollo de fármacos, tratos, y materiales orgánicos, incluidos los órganos artificiales. La nueva herramienta presentada por los autores podría significar un progreso 50 veces más rápido en estas áreas.














