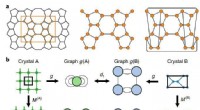
Diseñar nuevos compuestos o aleaciones cuyas superficies puedan usarse como catalizadores en reacciones químicas puede ser un proceso complejo que depende en gran medida de la intuición de químicos experimentados. Un equipo de investigadores del MIT ha ideado un nuevo enfoque que utiliza el aprendizaje automático, que elimina la necesidad de la intuición y proporciona información más detallada que la que los métodos convencionales pueden lograr en la práctica.
Por ejemplo, aplicando el nuevo sistema a un material que ya ha sido estudiado durante 30 años por medios convencionales, el equipo descubrió que la superficie del compuesto podría formar dos nuevas configuraciones atómicas que no habían sido identificadas previamente, y otra configuración vista en trabajos anteriores. Es probable que sea inestable.
Los hallazgos se describen en la revista Nature Computational Science. , en un artículo del estudiante graduado del MIT Xiaochen Du, los profesores Rafael Gómez-Bombarelli y Bilge Yildiz, el miembro del personal técnico del Laboratorio Lincoln del MIT, Lin Li, y otras tres personas.
Las superficies de los materiales a menudo interactúan con su entorno de maneras que dependen de la configuración exacta de los átomos en la superficie, que puede diferir según las partes de la estructura atómica del material que estén expuestas. Piense en un pastel de capas con pasas y nueces:dependiendo exactamente de cómo corte el pastel, diferentes cantidades y disposiciones de las capas y frutas quedarán expuestas en el borde de la rebanada.
El medio ambiente también importa. La superficie del pastel se verá diferente si se empapa en almíbar, lo que lo vuelve húmedo y pegajoso, o si se mete en el horno, lo que hace que la superficie quede crujiente y oscura. Esto es similar a cómo responden las superficies de los materiales cuando se sumergen en un líquido o se exponen a diferentes temperaturas.
Los métodos utilizados habitualmente para caracterizar las superficies de los materiales son estáticos y analizan una configuración particular entre millones de posibilidades. El nuevo método permite una estimación de todas las variaciones, basándose sólo en unos pocos cálculos de primeros principios elegidos automáticamente mediante un proceso iterativo de aprendizaje automático, para encontrar aquellos materiales con las propiedades deseadas.
Además, a diferencia de los métodos actuales típicos, el nuevo sistema se puede ampliar para proporcionar información dinámica sobre cómo las propiedades de la superficie cambian con el tiempo en condiciones operativas, por ejemplo, mientras un catalizador promueve activamente una reacción química, o mientras un electrodo de batería se está cargando o descargando.
El método de los investigadores, al que llaman marco de reconstrucción automática de superficies, evita la necesidad de utilizar ejemplos de superficies cuidadosamente seleccionados para entrenar la red neuronal utilizada en la simulación. En cambio, comienza con un único ejemplo de una superficie de corte prístina, luego utiliza el aprendizaje activo combinado con un tipo de algoritmo de Monte-Carlo para seleccionar sitios para muestrear en esa superficie, evaluando los resultados de cada sitio de ejemplo para guiar la selección del siguiente. sitios.
Utilizando menos de 5.000 cálculos de primeros principios, de los millones de composiciones y configuraciones químicas posibles, el sistema puede obtener predicciones precisas de las energías superficiales en diversos potenciales químicos o eléctricos, informa el equipo.
"Estamos analizando la termodinámica", dice Du, "lo que significa que, bajo diferentes tipos de condiciones externas como presión, temperatura y potencial químico, que pueden estar relacionados con la concentración de un determinado elemento, [podemos investigar] qué ¿Cuál es la estructura más estable para la superficie?"
En principio, determinar las propiedades termodinámicas de la superficie de un material requiere conocer las energías superficiales en una disposición atómica única específica y luego determinar esas energías millones de veces para abarcar todas las variaciones posibles y capturar la dinámica de los procesos que tienen lugar. Si bien en teoría es posible hacer esto computacionalmente, "simplemente no es asequible" a una escala de laboratorio típica, afirma Gómez-Bombarelli.
Los investigadores han podido obtener buenos resultados examinando sólo unos pocos casos específicos, pero no son casos suficientes para proporcionar una imagen estadística real de las propiedades dinámicas implicadas, afirma.
Utilizando su método, Du dice:"Tenemos nuevas características que nos permiten tomar muestras de la termodinámica de diferentes composiciones y configuraciones. También demostramos que podemos lograrlas a un costo menor, con evaluaciones de energía de la mecánica cuántica menos costosas. Y podemos También podemos hacer esto con materiales más duros", incluidos los materiales de tres componentes.
"Lo que tradicionalmente se hace en el campo", dice, "es que los investigadores, basándose en su intuición y conocimiento, probarán sólo unas pocas superficies de conjetura. Pero hacemos un muestreo completo y se hace automáticamente". Dice que "hemos transformado un proceso que alguna vez fue imposible o extremadamente desafiante debido a la necesidad de la intuición humana. Ahora, requerimos una participación humana mínima. Simplemente proporcionamos la superficie prístina y nuestra herramienta se encarga del resto". P>
Esa herramienta, o conjunto de algoritmos informáticos, llamado AutoSurfRecon, ha sido puesto a disposición de los investigadores de forma gratuita para que pueda ser descargado y utilizado por cualquier investigador del mundo para ayudar, por ejemplo, a desarrollar nuevos materiales para catalizadores, como por ejemplo el producción de hidrógeno "verde" como combustible alternativo libre de emisiones, o para nuevos componentes de baterías o pilas de combustible.
Por ejemplo, dice Gómez-Bombarelli, al desarrollar catalizadores para la producción de hidrógeno, "parte del problema es que no se entiende realmente en qué se diferencia su superficie de su volumen a medida que ocurre el ciclo catalítico. Entonces, existe esta desconexión entre la apariencia del material cómo cuándo se utiliza y cómo se ve cuando se prepara antes de ponerse en acción".
Añade que "al final del día, en catálisis, la entidad responsable de que el catalizador haga algo son unos pocos átomos expuestos en la superficie, por lo que realmente importa mucho cómo se ve exactamente la superficie en ese momento". P>
Otra aplicación potencial es el estudio de la dinámica de las reacciones químicas utilizadas para eliminar el dióxido de carbono del aire o de las emisiones de las centrales eléctricas. Estas reacciones a menudo funcionan mediante el uso de un material que actúa como una especie de esponja para absorber oxígeno, por lo que elimina los átomos de oxígeno de las moléculas de dióxido de carbono, dejando atrás monóxido de carbono, que puede ser un combustible útil o una materia prima química. Desarrollar este tipo de materiales "requiere comprender qué hace la superficie con los oxígenos y cómo está estructurada", afirma Gómez-Bombarelli.
Utilizando su herramienta, los investigadores estudiaron la disposición atómica de la superficie del material de perovskita, óxido de titanio y estroncio, o SrTiO3. , que ya había sido analizado por otros utilizando métodos convencionales durante más de tres décadas pero aún no se entendía completamente. Descubrieron dos nuevas disposiciones de los átomos en su superficie que no se habían informado previamente, y predicen que, de hecho, es poco probable que ocurra una disposición de la que se había informado.
"Esto pone de relieve que el método funciona sin intuiciones", afirma Gómez-Bombarelli. "Y eso es bueno porque a veces la intuición es errónea y lo que la gente pensaba que era cierto resulta no serlo". Esta nueva herramienta, afirmó, permitirá a los investigadores ser más exploratorios y probar una gama más amplia de posibilidades.
Ahora que su código ha sido lanzado a la comunidad en general, dice, "esperamos que sirva de inspiración para mejoras muy rápidas" por parte de otros usuarios.
El equipo incluía a James Damewood, Ph.D. estudiante del MIT, Jaclyn Lunger Ph.D., que ahora está en Flagship Pioneering, y Reisel Millan, un ex postdoctorado que ahora trabaja en el Instituto de Tecnología Química de España.
Más información: Simulaciones aceleradas por aprendizaje automático para permitir la reconstrucción de superficies sin heurísticas, Ciencia Computacional de la Naturaleza (2023). DOI:10.1038/s43588-023-00571-7
Información de la revista: Ciencia Computacional de la Naturaleza
Proporcionado por el Instituto de Tecnología de Massachusetts
Esta historia se vuelve a publicar por cortesía de MIT News (web.mit.edu/newsoffice/), un sitio popular que cubre noticias sobre investigación, innovación y enseñanza del MIT.