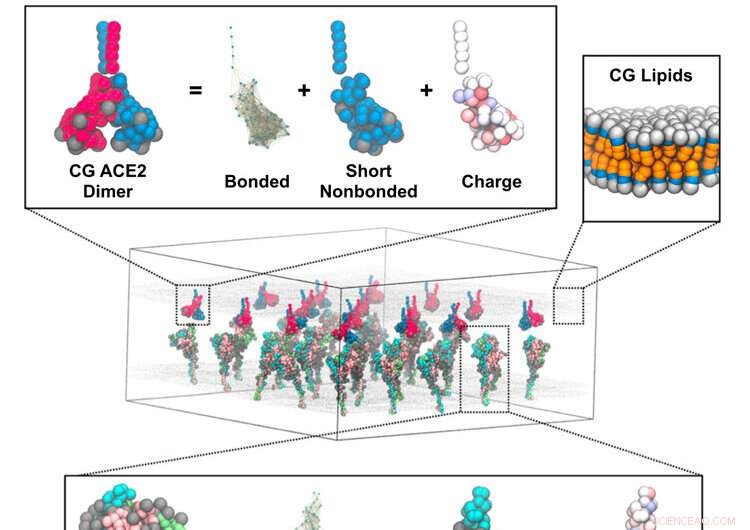
El mecanismo por el cual el coronavirus se fusiona con las células huésped ha sido sugerido a través de simulaciones realizadas por investigadores de la Universidad de Chicago que utilizan la supercomputadora Frontera en TACC. Representación representativa de una simulación de grano grueso (CG) de trímeros de punta en la membrana que interactúan con una membrana adyacente con dímeros ACE2. Los recuadros representan los componentes del modelo CG para el trímero de punta (abajo), el dímero ACE2 (arriba a la izquierda) y la membrana lipídica (arriba a la derecha). Crédito:Pak, A.J., Yu, A., Ke, Z. et al.
El misterio de cómo exactamente el virus SARS-CoV-2 infecta las células pulmonares humanas permanece en gran parte oculto para los científicos experimentales. Ahora, sin embargo, los detalles diabólicos del mecanismo por el cual el coronavirus se fusiona con las células huésped han sido sugeridos a través de simulaciones realizadas por investigadores de la Universidad de Chicago que utilizan la supercomputadora Frontera en el Centro de Computación Avanzada de Texas (TACC).
Los modelos informáticos muestran el comportamiento cooperativo de las proteínas receptoras de la célula huésped que conduce a su propia infección. El trabajo se puede aplicar para ayudar a comprender la mayor virulencia de las variantes de coronavirus como delta, omicron y más.
"Descubrimos que la proteína espiga interactúa con dos receptores ACE2 de una manera muy cooperativa", dijo Gregory Voth, distinguido profesor de química de la Universidad de Chicago. "Esta es una idea biofísica fundamental".
Voth es el autor principal del estudio que modeló las interacciones entre el coronavirus y las células receptoras con simulaciones por computadora publicado en la revista Nature Communications en febrero de 2022.
Como un balón de fútbol con púas, las proteínas de las púas adornan la superficie del coronavirus. Los picos buscan y se fusionan con los receptores de la proteína de la enzima convertidora de angiotensina 2 (ACE2) en las células pulmonares humanas. La proteína espiga se compone de dos partes principales. El dominio S1 contiene el dominio de unión al receptor que reconoce las proteínas ACE2. Y el dominio S2 contiene la maquinaria de fusión, que está protegida y cubierta como una cáscara por el dominio S1.
Las simulaciones revelan cómo una proteína del receptor ACE2 se aferra al pico de coronavirus y lo debilita mientras que la otra comienza a separarlo. El dominio S1 luego se desmorona y expone la maquinaria de fusión. Este golpe "uno-dos" prepara al virus para la fusión y la entrada en las células huésped del pulmón humano.
"Parece que las variantes como delta y omicron pueden acentuar ese comportamiento aún más, es un paso clave. En última instancia, los futuros anticuerpos y posiblemente los productos farmacéuticos moleculares deberían poder interferir con este proceso", dijo Voth.
Voth y sus colegas desarrollaron lo que ellos llaman "modelos de grano grueso de abajo hacia arriba" que tomaron datos de tomografía crioelectrónica del laboratorio del coautor del estudio John Briggs del Instituto Max Planck de Bioquímica. Lo combinaron con simulaciones de dinámica molecular atomística. Los datos generados alimentaron un marco teórico que desarrolló los modelos de grano grueso.
"Los modelos de grano grueso son hasta 1000 veces más rápidos que las simulaciones de dinámica molecular atomística directa, pero conservan las características físicas esenciales", dijo Voth. Este método proporciona un enorme ahorro de tiempo y dinero en los cálculos.
El equipo científico recibió recursos y servicios de supercomputadoras del Consorcio HPC COVID-19, un esfuerzo público-privado en apoyo de la investigación COVID-19. A través del consorcio, utilizaron el sistema Frontera financiado por la Fundación Nacional de Ciencias en TACC; el clúster de computadoras Witherspoon en IBM Research; y recursos de Oak Ridge Leadership Computing Facility en el Laboratorio Nacional de Oak Ridge.
"Calculamos datos de dinámica molecular de todos los átomos en Frontera y usamos herramientas de análisis disponibles de TACC, ambas fueron muy valiosas", dijo Voth.
El equipo de Voth presentó su artículo antes de que se conocieran las variantes delta y omicron y, por lo tanto, no predijo las mutaciones. Pero volvieron y revisaron los modelos para investigar las variantes.
"Delta tiene algo así como una apertura en el pico que ocurre más fácilmente que en mutaciones anteriores de coronavirus", dijo Voth. "Fue emocionante desde un punto de vista científico ver comportamientos que no se habían visto antes".
Voth se refirió a datos de laboratorio de microscopía crioelectrónica que mostraban la estructura de una proteína espiga soluble con dos receptores ACE2 unidos a ella. Pero distinguió este ejemplo cristalizado de lo que investigó utilizando simulaciones en el entorno más realista de muchas proteínas que interactúan entre sí en láminas de membrana.
"Las supercomputadoras, si se usan bien y se basan en buena física, pueden proporcionar una forma completamente nueva de ver estos procesos. A través de la simulación por computadora, uno puede estudiar cosas que actualmente no se pueden hacer con experimentos. La simulación y los experimentos funcionan muy bien juntos, de la mano", dijo Voth. El primer modelo completo de coronavirus muestra cooperación