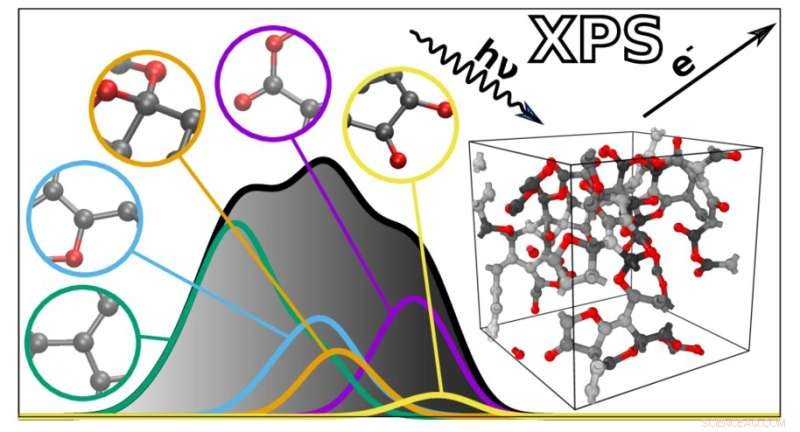
El nuevo algoritmo predice los espectros XPS de materiales complejos en función de las contribuciones atómicas individuales. Crédito:Miguel Caro / Universidad Aalto
Los materiales a base de carbono tienen un enorme potencial para construir un futuro sostenible, pero los científicos de materiales necesitan herramientas para analizar adecuadamente su estructura atómica, que determina sus propiedades funcionales. La espectroscopia de fotoelectrones de rayos X (XPS) es una de las herramientas utilizadas para hacer esto, pero los resultados de XPS pueden ser difíciles de interpretar. Ahora, los investigadores de Aalto han desarrollado una herramienta de aprendizaje automático para mejorar los análisis XPS, que han puesto a disposición de forma gratuita como XPS Prediction Server.
Los espectros XPS son gráficos con una colección de picos que reflejan la energía de enlace de los electrones en lo profundo de los átomos que forman un material. Debido a que las energías de enlace dependen del entorno atómico, se pueden usar para inferir cómo están conectados los átomos en un material o molécula en particular. Sin embargo, esto también hace que los espectros XPS sean difíciles de interpretar, ya que muchos factores afectan las energías de enlace. Las energías de enlace de diferentes características atómicas también pueden superponerse, lo que complica aún más el análisis.
Para ayudar con esto, un equipo dirigido por Miguel Caro desarrolló un método computacional que puede predecir el espectro de energía de enlace de un material basado en un modelo estructural generado por computadora. Esto simplifica la interpretación de datos XPS al hacer posible comparar las energías de enlace observadas experimentalmente con las predicciones computacionales.
La idea en sí no es nueva, pero el problema ha sido la dificultad computacional de calcular con precisión el espectro XPS de un material. El equipo de Caro resolvió esto usando aprendizaje automático. El truco consistía en entrenar un algoritmo informático económico para predecir el resultado de un método de referencia computacionalmente costoso basado en una combinación eficiente de datos mecánicos cuánticos computacionalmente baratos y costosos.
El método computacionalmente más barato, DFT, no coincide con los resultados experimentales con mucha precisión. El método más preciso, GW, tarda demasiado en calcularse cuando una molécula tiene muchos átomos. "Decidimos construir un modelo de referencia que utiliza abundantes datos DFT y luego refinarlo con datos GW escasos y valiosos. Y funcionó", dice Caro.
El algoritmo resultante puede predecir el espectro de cualquier material desordenado hecho de carbono, hidrógeno y oxígeno. "Los espectros predichos son notablemente similares a los obtenidos experimentalmente. Esto abre la puerta a una mejor integración entre la caracterización experimental y computacional de los materiales", dice Caro. A continuación, el equipo planea ampliar su técnica para incluir una gama más amplia de materiales y otros tipos de espectroscopia.
El artículo de acceso abierto se publicó en Chemistry of Materials . El carbono tipo diamante se forma de manera diferente a lo que se creía:el aprendizaje automático permite el desarrollo de un nuevo modelo