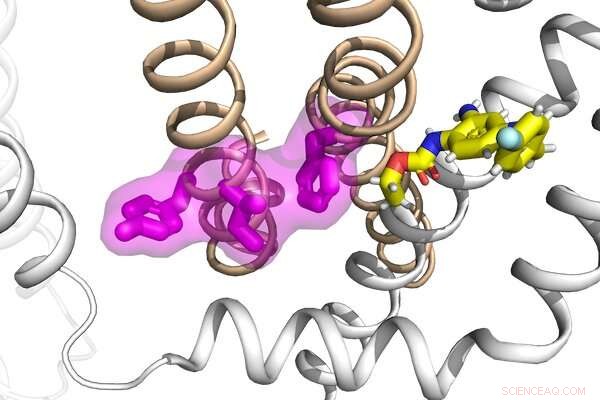
Los investigadores del laboratorio de Jianmin Cui han analizado los mecanismos detrás de la función y la disfunción de un grupo de proteínas, así como sus interacciones con un fármaco antiepiléptico, para desarrollar una nueva estrategia potencial para tratar la epilepsia. Crédito:laboratorio Cui
La epilepsia es un trastorno neurológico que surge de una actividad eléctrica anormal en el cerebro que conduce a convulsiones. Estos eventos convulsivos pueden tener una variedad de causas, incluidas variantes genéticas en una familia de proteínas que regulan los iones de potasio en el cerebro. Investigadores de la Universidad de Washington en St. Louis han dirigido un equipo internacional para observar de cerca los mecanismos detrás de la función y la disfunción de estas proteínas, así como sus interacciones con un fármaco antiepiléptico, para desarrollar una nueva estrategia potencial para tratar la epilepsia.
Jianmin Cui, profesor de ingeniería biomédica en la Escuela de Ingeniería McKelvey, y Nien-Du Yang, estudiante de doctorado en ingeniería biomédica que realiza investigaciones en el laboratorio de Cui, se asociaron con Harley Kurata, profesor asociado de farmacología en la Universidad de Alberta, y investigó el mecanismo de trabajo de dos canales de iones de potasio, KCNQ2 y KCNQ3. Sus hallazgos descubren un mecanismo conservado para la activación del canal KCNQ que es un objetivo tanto de las mutaciones vinculadas a la epilepsia como de un compuesto de molécula pequeña.
El trabajo fue publicado el 20 de julio en Science Advances .
La familia de canales de potasio KCNQ tiene múltiples funciones, desde regular los latidos del corazón (mediante KCNQ1) hasta controlar la excitabilidad de las neuronas (mediante KCNQ2-5). Estos canales se activan por voltaje para que detecten cambios de voltaje a través de la membrana celular y se abran y cierren en respuesta. La comunicación entre la detección de voltaje y la apertura de los poros del canal se conoce como acoplamiento electromecánico, un proceso que implica cambios conformacionales de la proteína durante la activación dependiente del voltaje.
El equipo de Cui ha demostrado previamente que KCNQ1, la isoforma cardíaca de KCNQ, presenta un proceso de dos etapas en el acoplamiento electromecánico que conduce a dos estados abiertos de canal distintos, el intermedio abierto y el abierto activado. La regulación de los dos estados abiertos subyace a las modulaciones específicas de tejido, la patogénesis de la enfermedad y la farmacología de KCNQ1. KCNQ2 y KCNQ3 se expresan en gran medida en el sistema nervioso central y son los principales contribuyentes a la corriente M, una corriente de potasio fundamental que modula la excitabilidad neuronal. Por lo tanto, el deterioro de la función de corriente M por mutaciones congénitas en KCNQ2 y KCNQ3 se asocia comúnmente con epilepsia pediátrica y de inicio temprano.
"Aunque los canales KCNQ son muy similares en sus secuencias y estructuras, no está claro si las isoformas neuronales KCNQ también comparten el mismo mecanismo de acoplamiento electromecánico o dos estados abiertos", dijo Yang, el primer autor del artículo. "Este trabajo revela similitudes y diferencias clave entre estos canales que pueden tener implicaciones importantes para su función en los cardiomiocitos o las neuronas".
El equipo utilizó una variedad de métodos para estudiar el mecanismo de acoplamiento electromecánico en estos canales de potasio, incluida la creación de mutaciones genéticas específicas en los canales, electrofisiología y mediciones ópticas de fluorescencia.
"Elucidar el mecanismo molecular para el acoplamiento electromecánico es un paso importante hacia la comprensión de la activación de los canales de potasio dependiente del voltaje", dijo Cui. "Proporcionamos evidencia funcional de que los canales neuronales KCNQ2 y KCNQ3 son diferentes de KCNQ1 en el sentido de que presentan un solo estado activado-abierto pero con un mecanismo de acoplamiento electromecánico conservado específico para el estado activado-abierto".
Estos canales son objetivos principales para los tratamientos para la epilepsia, encontraron los investigadores. El equipo también identificó un conjunto de mutaciones en KCNQ2 y KCNQ3 asociadas con la encefalopatía epiléptica infantil temprana, una forma grave de epilepsia infantil, que interrumpe específicamente el acoplamiento electromecánico de los canales. Los investigadores aprovecharon un prototipo de fármaco antiepiléptico, la retigabina, dado su mecanismo de acción en los canales KCNQ neuronales, y demostraron que el acoplamiento electromecánico se puede mejorar directamente para rescatar la función de estos mutantes enfermos. Sus estudios sugieren que el mecanismo de acoplamiento electromecánico en los canales KCNQ puede ser un objetivo eficaz, presentando una nueva estrategia farmacológica para desarrollar terapias más eficaces para el tratamiento de la epilepsia. Disfunción de los canales de potasio en la epilepsia genética