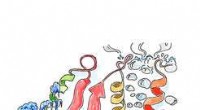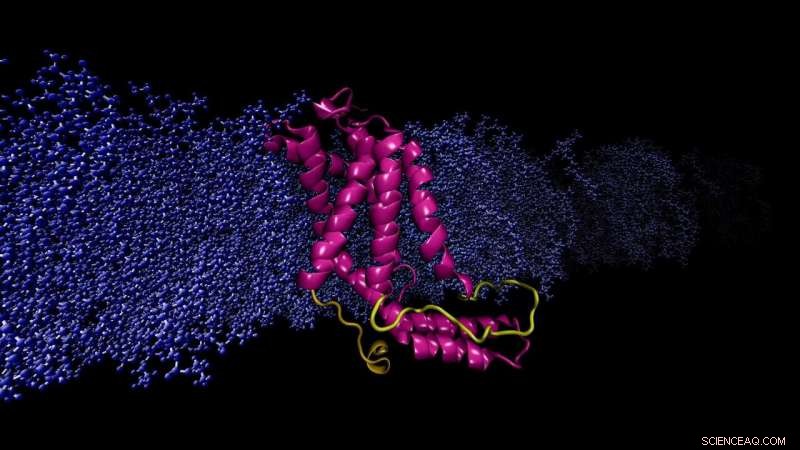
Modelo computacional de membrana celular incrustada en la proteína YidC2. El bucle modelado (amarillo), falta en la estructura del cristal de rayos X, es crucial para la estabilización de proteínas. Crédito:Sogol Moradi
Un nuevo estudio realizado por químicos de la Universidad de Arkansas muestra que la cristalografía de rayos X, el método estándar para determinar la estructura de las proteínas, puede proporcionar información inexacta sobre un conjunto crítico de proteínas, las que se encuentran en las membranas celulares, lo que a su vez podría dar lugar a un diseño de fármacos deficiente e ineficaz.
Los hallazgos de los investigadores se publicaron hoy en Informes científicos , una publicación de Nature.
"Dos tercios de todas las drogas, incluidos los utilizados para la quimioterapia, proteínas diana que se encuentran en las membranas celulares, "dijo Mahmoud Moradi, profesor asistente de química y bioquímica en la Facultad de Artes y Ciencias J. William Fulbright. "Desafortunadamente, Cristalografía de rayos X, el estándar de oro para determinar la estructura de las proteínas, tiene muchas limitaciones cuando se trata de las que se encuentran en la membrana celular. Nuestro trabajo expone, y de muchas formas explica estas limitaciones ".
Considerado el caballo de batalla de las moléculas de las células, las proteínas son responsables de casi todas las tareas de los sistemas vivos. Algunas proteínas viven dentro de las células, y algunos residen en la membrana celular, una capa externa de lípidos que separa la célula de su entorno externo. Las proteínas de membrana son de vital importancia porque regulan el intercambio de información y materiales entre la célula y su entorno. una tarea vital para la supervivencia y el funcionamiento normal de la célula porque cualquier trastorno en la función de las proteínas puede provocar una enfermedad.
El estudio de la función de las proteínas es necesario para comprender la base molecular de la enfermedad. Para hacer esto, los investigadores se han basado en la cristalografía de rayos X, la herramienta principal para determinar la forma y estructura de las proteínas. La cristalografía de rayos X también es fundamental para el diseño de fármacos que manipulen eficazmente la función de las proteínas. Sin embargo, el estudio de la estructura de las proteínas de la membrana es difícil porque su entorno nativo no es compatible con la cristalografía de rayos X. Los investigadores deben eliminar las proteínas de su entorno nativo y colocarlas en un entorno lipídico artificial antes de aplicar la técnica.
Moradi y Thomas Harkey, un estudiante de pregrado en ese momento y ahora estudiante de medicina en la Universidad de Arkansas para Ciencias Médicas, abordaron este problema desde un ángulo diferente. Durante aproximadamente dos años, utilizaron una supercomputadora en el Arkansas High Performance Computing Center para funcionar de manera continua, cálculos a nivel de microsegundos que simulan la dinámica molecular de YidC2, una proteína de membrana con un bucle citoplasmático sin resolver cristalográficamente en su estructura molecular. Se sabe que los bucles citoplasmáticos tienen importancia funcional en las proteínas de membrana.
Las simulaciones de Moradi y Harkey demostraron que el bucle citoplásmico de YidC2 estabilizó la proteína completa, particularmente la región C1, un área potencialmente importante para el diseño de fármacos. Los grupos de cabeza de lípidos altamente polares o cargados interactuaron con el asa y lo estabilizaron. Este hallazgo demostró que los bucles no resueltos de proteínas de membrana podrían ser importantes para la estabilización de proteínas, a pesar de la aparente falta de estructura molecular.
"Típicamente, si parte de una proteína no se resuelve en cristalografía de rayos X, se interpreta como carente de una estructura particular, ", Dijo Moradi." Demostramos que para las proteínas de la membrana y, en particular, las partes de la proteína que interactúan con la membrana celular, esta interpretación no es precisa y podría inducir a error. Creemos que la explicación alternativa para el trastorno podría ser que la proteína no se estudia en su entorno de membrana nativo ".
Moradi dijo que sus resultados también demostraron que la química computacional y la tecnología de supercomputación se pueden utilizar para modelar proteínas de membrana con mayor precisión en un entorno que imita su entorno fisiológico.