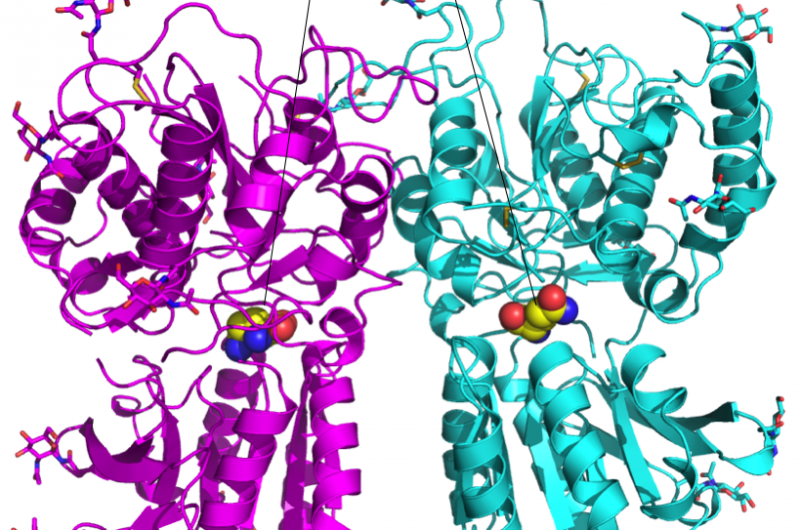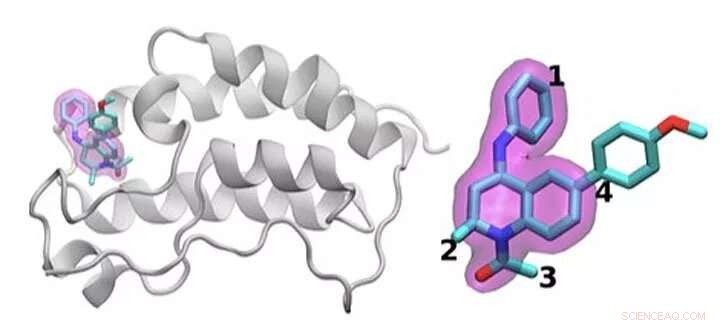
Un esquema de la proteína BRD4 unida a uno de los 16 fármacos basados en el mismo andamio de tetrahidroquinolina (resaltado en magenta). Las regiones que se modifican químicamente entre los medicamentos investigados en este estudio se etiquetan del 1 al 4. Por lo general, sólo se realiza un pequeño cambio en la estructura química de un fármaco al siguiente. Este enfoque conservador permite a los investigadores explorar por qué un fármaco es eficaz y otro no. Crédito:Laboratorio Nacional Brookhaven
Identificar el tratamiento farmacológico óptimo es como alcanzar un objetivo en movimiento. Para detener la enfermedad Los medicamentos de molécula pequeña se unen estrechamente a una proteína importante, bloqueando sus efectos en el cuerpo. Incluso los medicamentos aprobados no suelen funcionar en todos los pacientes. Y con el tiempo los agentes infecciosos o las células cancerosas pueden mutar, inutilizando una droga que alguna vez fue eficaz.
Un problema físico fundamental subyace a todas estas cuestiones:optimizar la interacción entre la molécula del fármaco y su objetivo proteico. Las variaciones en las moléculas candidatas a fármacos, el rango de mutación en las proteínas y la complejidad general de estas interacciones físicas dificultan este trabajo.
Shantenu Jha, del Laboratorio Nacional Brookhaven del Departamento de Energía (DOE) y de la Universidad de Rutgers, dirige un equipo que intenta simplificar los métodos computacionales para que las supercomputadoras puedan asumir parte de esta inmensa carga de trabajo. Han encontrado una nueva estrategia para abordar una parte:diferenciar cómo los candidatos a fármacos interactúan y se unen a una proteína objetivo.
Por su trabajo, Jha y sus colegas ganaron el premio IEEE International Scalable Computing Challenge (SCALE) del año pasado, que reconoce soluciones informáticas escalables para problemas de ciencia e ingeniería del mundo real.
Para diseñar un nuevo fármaco, una compañía farmacéutica puede comenzar con una biblioteca de millones de moléculas candidatas que reducen a miles que muestran alguna unión inicial a una proteína objetivo. El perfeccionamiento de estas opciones para obtener un fármaco útil que se pueda probar en humanos puede implicar experimentos extensos para sumar o restar grupos de átomos en ubicaciones clave de la molécula y probar cómo cada uno de estos cambios altera la forma en que interactúan la molécula pequeña y la proteína.
Las simulaciones pueden ayudar con este proceso. Más grande, Las supercomputadoras más rápidas y los algoritmos cada vez más sofisticados pueden incorporar física realista y calcular las energías de enlace entre varias moléculas pequeñas y proteínas. Estos métodos pueden consumir importantes recursos computacionales, sin embargo, para lograr la precisión necesaria. Las simulaciones útiles para la industria también deben proporcionar respuestas rápidas. Debido al tira y afloja entre precisión y velocidad, los investigadores están innovando constantemente, desarrollar algoritmos más eficientes y mejorar el rendimiento, Dice Jha.
Este problema también requiere administrar los recursos computacionales de manera diferente que para muchos otros problemas a gran escala. En lugar de diseñar una única simulación que se escale para utilizar una supercomputadora completa, Los investigadores ejecutan simultáneamente muchos modelos más pequeños que se moldean entre sí y la trayectoria de los cálculos futuros, una estrategia conocida como computación basada en conjuntos, o flujos de trabajo complejos.
"Piense en esto como intentar explorar un paisaje abierto muy grande para tratar de encontrar dónde podría obtener el mejor candidato a fármaco, "Jha dice. En el pasado, Los investigadores han pedido a las computadoras que naveguen por este panorama haciendo elecciones estadísticas aleatorias. En un punto de decisión, la mitad de los cálculos pueden seguir un camino, la otra mitad otra.
Jha y su equipo buscan formas de ayudar a estas simulaciones a aprender del paisaje. Ingerir y luego compartir datos en tiempo real no es fácil, Jha dice:"y eso es lo que requirió que algunas de las innovaciones tecnológicas se hicieran a escala". Él y su equipo de Rutgers están colaborando con el grupo de Peter Coveney en el University College London en este trabajo.
Para probar esta idea, han utilizado algoritmos que predicen la afinidad de enlace y han introducido versiones optimizadas en un marco HTBAC, para calculadora de afinidad de enlace de alto rendimiento. Una de esas calculadoras, conocido como ESMACS, les ayuda a eliminar las moléculas que se unen mal a una proteína objetivo. El otro, CORBATAS, es más precisa pero de alcance más limitado y requiere 2,5 veces más recursos computacionales. Sin embargo, puede ayudar a los investigadores a optimizar una interacción prometedora entre un fármaco y una proteína. El marco HTBAC les ayuda a implementar estos algoritmos de manera eficiente, guardando el algoritmo más intensivo para situaciones en las que sea necesario.
El equipo demostró la idea examinando 16 candidatos a fármacos de una biblioteca de moléculas en GlaxoSmithKline (GSK) con su objetivo, BRD4-BD1:una proteína que es importante en el cáncer de mama y las enfermedades inflamatorias. Los candidatos a fármacos tenían la misma estructura central pero diferían en cuatro áreas distintas alrededor de los bordes de la molécula.
En este estudio inicial, el equipo ejecutó miles de procesos simultáneamente en 32, 000 núcleos en aguas azules, una supercomputadora de la National Science Foundation (NSF) en la Universidad de Illinois en Urbana-Champaign. Han realizado cálculos similares en Titán, la supercomputadora Cray XK7 en Oak Ridge Leadership Computing Facility, una instalación para usuarios de la Oficina de Ciencias del DOE. El equipo distinguió con éxito entre la unión de estos 16 fármacos candidatos, la mayor simulación de este tipo hasta la fecha. "No solo alcanzamos una escala sin precedentes, "Dice Jha." Nuestro enfoque muestra la capacidad de diferenciar ".
Ganaron su premio SCALE por esta prueba de concepto inicial. El desafío ahora Jha dice:se asegura de que no solo funcione para BRD4, sino también para otras combinaciones de moléculas de fármacos y objetivos de proteínas.
Si los investigadores pueden seguir ampliando su enfoque, estas técnicas podrían eventualmente ayudar a acelerar el descubrimiento de fármacos y permitir la medicina personalizada. Pero para examinar problemas más realistas, necesitarán más potencia computacional. "Estamos en medio de esta tensión entre un espacio químico muy grande que nosotros, en principio, Necesito explorar y, lamentablemente recursos informáticos limitados ", dice Jha.
Incluso cuando la supercomputación se expande hacia la exaescala, Los científicos computacionales pueden más que llenar el vacío agregando física más realista a sus modelos. En el futuro inmediato, los investigadores deberán ser ingeniosos para ampliar estos cálculos. La necesidad es la madre de la innovación, Jha dice:precisamente porque la ciencia molecular no dispondrá de la cantidad ideal de recursos computacionales para realizar simulaciones.
Pero la computación a exaescala puede ayudarlos a acercarse a sus objetivos. Además de trabajar con University College London y GSK, Jha y sus colegas están colaborando con Rick Stevens de Argonne National Laboratory y el equipo de CANcer Distributed Learning Environment (CANDLE). Este proyecto de codiseño dentro del Proyecto de Computación Exascale del DOE está construyendo redes neuronales profundas y técnicas generales de aprendizaje automático para estudiar el cáncer. Los algoritmos y el software dentro de HTBAC podrían complementar el enfoque de CANDLE en esos enfoques.
Esta colaboración más amplia entre el grupo de Jha, el equipo de CANDLE y el laboratorio de John Chodera en el Memorial Sloan-Kettering Cancer Center han llevado al proyecto de Predicción Integrada y Escalable de Resistencia (INSPIRE). Este equipo ya ha realizado simulaciones en la supercomputadora Summit del DOE en el Laboratorio Nacional de Oak Ridge. Pronto continuará este trabajo en Frontera, la máquina de liderazgo de la NSF en el Centro de Computación Avanzada de Texas de la Universidad de Texas en Austin.
"Estamos ansiosos por un mayor progreso y mayores mejoras metodológicas, "Dice Jha." Nos gustaría ver cómo estos enfoques bastante complementarios podrían funcionar de manera integradora hacia esta gran visión ".