Crédito:Michele Ceriotti / EPFL
Hoy en día, muchas drogas se producen en forma de polvo sólido. Pero para comprender completamente cómo se comportarán los ingredientes activos una vez dentro del cuerpo, los científicos necesitan conocer su estructura exacta a nivel atómico. Por ejemplo, la forma en que las moléculas están dispuestas dentro de un cristal tiene un impacto directo en las propiedades de un compuesto, como su solubilidad. Por lo tanto, los investigadores están trabajando arduamente para desarrollar tecnologías que puedan identificar fácilmente las estructuras cristalinas exactas de los polvos microcristalinos.
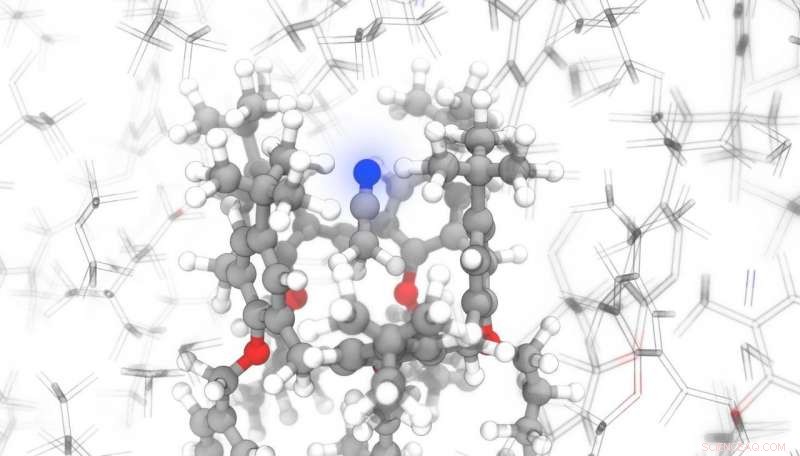
Un equipo de científicos de la EPFL ha escrito un programa de aprendizaje automático que puede predecir, en tiempo récord, cómo responderán los átomos a un campo magnético aplicado. Esto se puede combinar con espectroscopía de resonancia magnética nuclear (RMN) para determinar la ubicación exacta de los átomos en compuestos orgánicos complejos. Esto puede ser de gran beneficio para las empresas farmacéuticas. que deben monitorear cuidadosamente las estructuras de sus moléculas para cumplir con los requisitos de seguridad del paciente. Su investigación ha sido publicada en Comunicaciones de la naturaleza .
Velocidades vertiginosas con IA
La espectroscopía de RMN es un método bien conocido y altamente eficiente para sondear los campos magnéticos entre átomos y determinar cómo los átomos vecinos interactúan entre sí. Sin embargo, La determinación de la estructura cristalina completa por espectroscopia de RMN requiere un proceso extremadamente complicado, cálculos que requieren mucho tiempo y que involucran química cuántica, casi imposible para moléculas con estructuras muy intrincadas.
Pero el programa desarrollado en EPFL puede superar estos obstáculos. Los científicos entrenaron su modelo de IA en estructuras moleculares tomadas de bases de datos estructurales. "Incluso para moléculas relativamente simples, este modelo es casi 10, 000 veces más rápido que los métodos existentes, y la ventaja crece enormemente cuando se consideran compuestos más complejos, "dice Michele Ceriotti, jefe del Laboratorio de Ciencias Computacionales y Modelado de la Escuela de Ingeniería de EPFL y coautor del estudio. "Para predecir la firma de RMN de un cristal con casi 1, 600 átomos, nuestra técnica, ShiftML, requiere unos seis minutos; la misma hazaña habría llevado 16 años con técnicas convencionales ".
Este nuevo programa permitirá utilizar enfoques completamente diferentes que serán más rápidos y permitirán el acceso a moléculas más grandes. "Esto es realmente emocionante porque la aceleración masiva en los tiempos de cálculo nos permitirá cubrir espacios conformacionales mucho más grandes y determinar correctamente estructuras donde antes no era posible. Esto pone a la mayoría de las complejas moléculas de fármacos contemporáneos al alcance de la mano". "dice Lyndon Emsley, jefe del Laboratorio de Resonancia Magnética de la Facultad de Ciencias Básicas de la EPFL y coautor del estudio.
El programa ahora está disponible gratuitamente en línea. "Cualquiera puede cargar una molécula y obtener su firma de RMN en solo unos minutos, "dice Ceriotti.