Las nanoestructuras de carbono podrían ser más fáciles de diseñar y sintetizar gracias a un método de aprendizaje automático que predice cómo crecen en superficies metálicas. El nuevo enfoque, desarrollado por investigadores de la Universidad Tohoku de Japón y la Universidad Jiao Tong de Shanghai de China, facilitará la explotación de la versatilidad química única de la nanotecnología de carbono. El método fue publicado en la revista Nature Communications. .
Se ha estudiado ampliamente el crecimiento de nanoestructuras de carbono en una variedad de superficies, incluidas películas atómicamente delgadas, pero se sabe poco sobre la dinámica y los factores a nivel atómico que gobiernan la calidad de los materiales resultantes. "Nuestro trabajo aborda un desafío crucial para aprovechar el potencial de las nanoestructuras de carbono en dispositivos electrónicos o de procesamiento de energía", afirma Hao Li, del equipo de la Universidad de Tohoku.
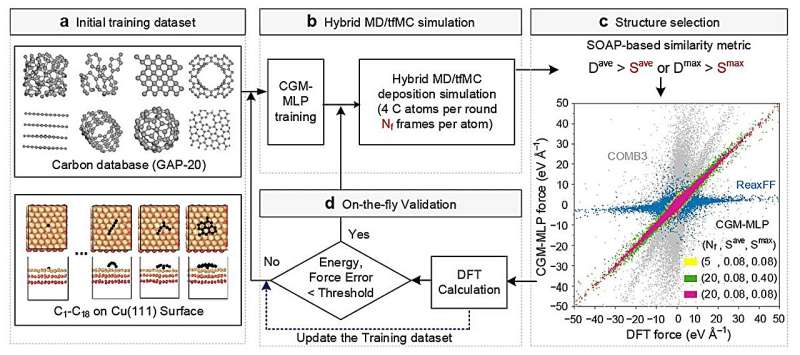
La amplia gama de superficies posibles y la sensibilidad del proceso a varias variables hacen que la investigación experimental directa sea un desafío. Por lo tanto, los investigadores recurrieron a las simulaciones de aprendizaje automático como una forma más eficaz de explorar estos sistemas.
Con el aprendizaje automático, se pueden combinar varios modelos teóricos con datos de experimentos químicos para predecir la dinámica del crecimiento cristalino de carbono y determinar cómo se puede controlar para lograr resultados específicos. El programa de simulación explora estrategias e identifica cuáles funcionan y cuáles no, sin necesidad de que humanos guíen cada paso del proceso.
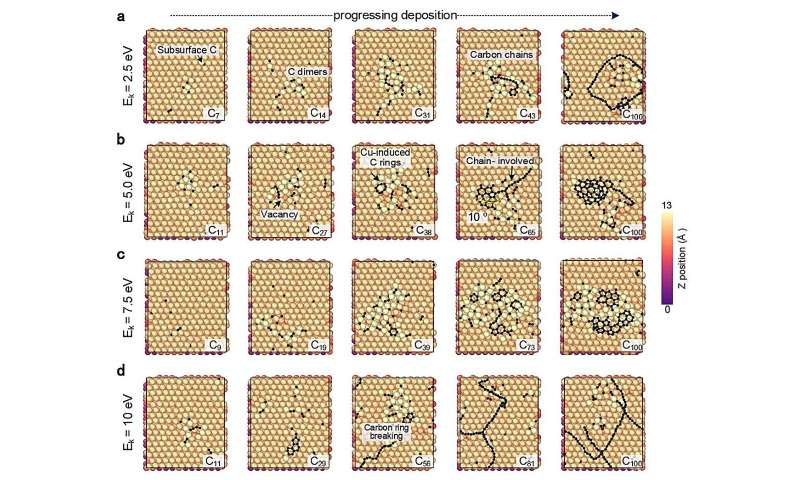
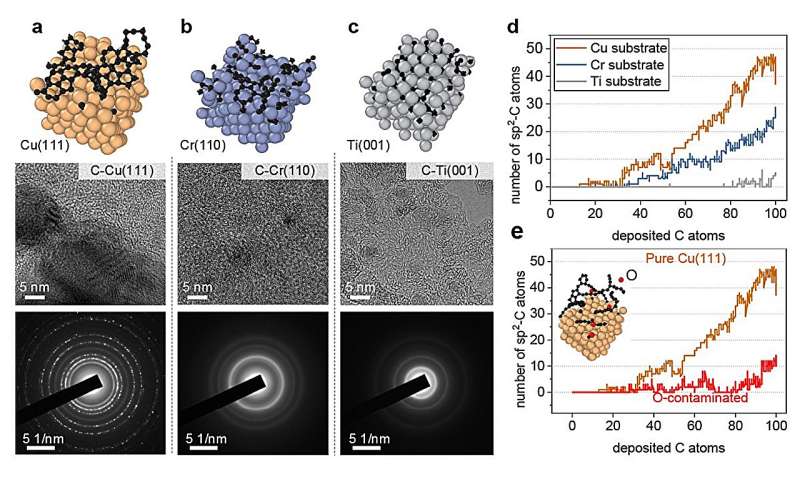
Los investigadores probaron este enfoque investigando simulaciones del crecimiento de grafeno, una forma de carbono, sobre una superficie de cobre. Después de establecer el marco básico, mostraron cómo su enfoque también podría aplicarse a otras superficies metálicas, como el titanio, el cromo y el cobre contaminados con oxígeno.
La distribución de electrones alrededor de los núcleos de los átomos en diferentes formas de cristales de grafeno puede variar. Estas sutiles diferencias en la estructura atómica y la disposición de los electrones afectan las propiedades químicas y electroquímicas generales del material. El enfoque de aprendizaje automático puede probar cómo estas diferencias afectan la difusión de átomos individuales y átomos unidos y la formación de cadenas de carbono, arcos y estructuras de anillos.
El equipo validó los resultados de las simulaciones mediante experimentos y descubrió que coincidían estrechamente. "En general, nuestro trabajo proporciona un método práctico y eficiente para diseñar sustratos metálicos o de aleaciones para lograr las nanoestructuras de carbono deseadas y explorar nuevas oportunidades", afirma Li.
Añade que el trabajo futuro se basará en esto para investigar temas como las interfaces entre sólidos y líquidos en catalizadores avanzados y las propiedades químicas de los materiales utilizados para procesar y almacenar energía.
Más información: Di Zhang et al, Modelo activo de aprendizaje automático para la simulación dinámica y los mecanismos de crecimiento del carbono en una superficie metálica, Nature Communications (2024). DOI:10.1038/s41467-023-44525-z
Proporcionado por la Universidad de Tohoku