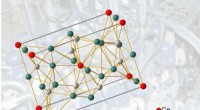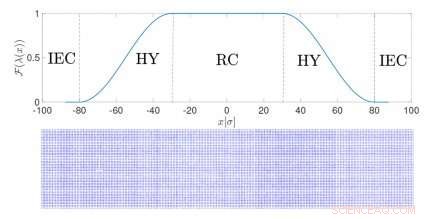
Configuración de una simulación de resolución adaptativa para sólidos. Crédito:Springer
Las simulaciones por computadora se utilizan para comprender las propiedades de la materia blanda, como líquidos, polímeros y biomoléculas como el ADN, que son demasiado complicados para ser descritos por ecuaciones. Suelen ser demasiado caras para simularlas en su totalidad, dada la potencia computacional intensiva requerida. En lugar de, una estrategia útil es acoplar un modelo preciso, aplicado en las áreas del sistema que requieren mayor atención, con un modelo más simple, modelo idealizado.
En un artículo reciente publicado en EPJ E , Maziar Heidari, del Instituto Max Planck de Investigación de Polímeros, Maguncia, Alemania y sus colegas logran que el modelo preciso en alta resolución coincida perfectamente con una representación que se pueda resolver exactamente a una resolución más baja.
El ideal, El modelo más simple es una especie de representación desnuda de átomos o moléculas, que no interactúan entre sí. Estudios anteriores han aplicado esta estrategia a líquidos, pero en este estudio, los autores lo aplican por primera vez a un modelo sólido acoplado a un cristal ideal, en el que los átomos tienen movimientos restringidos y no interactúan, apodado cristal de Einstein. El equipo pudo calcular sus propiedades termodinámicas, por ejemplo. temperatura y energía libre, a un costo computacional reducido.
En este tipo de simulación, llamadas simulaciones de resolución adaptativa, la resolución de una molécula depende de su posición en el espacio. En la región de transición entre las dos resoluciones, las moléculas se adaptan a un modelo u otro. Esta es una forma eficiente de calcular las características termodinámicas relevantes del sólido real descomponiéndolas en una contribución ideal (del modelo simplificado) y otro término, específico del sistema en particular. La metodología combina la simplicidad de los modelos ideales con la precisión química de las representaciones realistas.