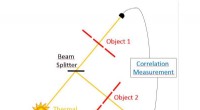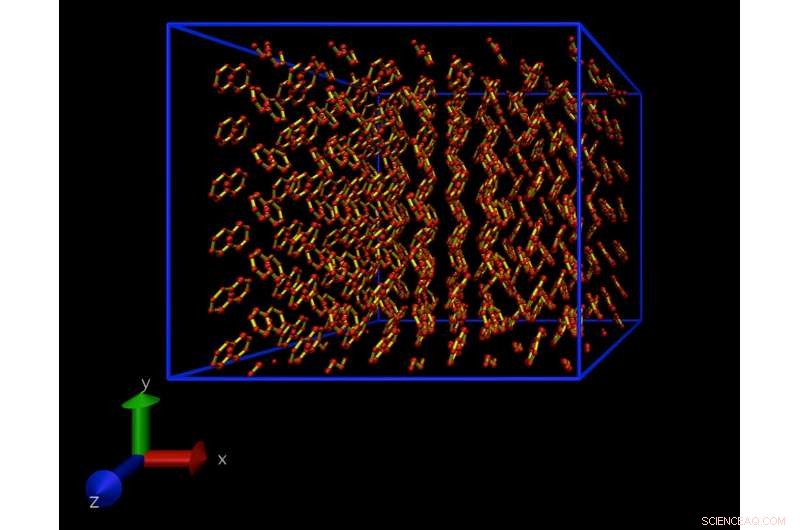
Una instantánea de una simulación de dinámica molecular de un modelo atomístico de un cristal de naftaleno. Este cristal se repite periódicamente en todas direcciones, para eliminar efectos superficiales. Crédito:Daan Frenkel, Universidad de Cambridge
La solubilidad de cualquier sustancia dada (la medida de qué tan bien se disuelve la sustancia en otra sustancia conocida como solvente) depende de propiedades básicas como la temperatura y la presión, así como las identidades químicas de la sustancia disuelta (el soluto) y el solvente.
Predecir la solubilidad es importante para una variedad de aplicaciones. En el campo farmacéutico, por ejemplo, es fundamental conocer la solubilidad de un fármaco, ya que determina directamente su disponibilidad para el organismo. La industria del petróleo ofrece otro ejemplo:las sustancias con baja solubilidad pueden formar incrustaciones o depósitos no deseados en tuberías o taladros. causando bloqueos y otros grandes problemas.
A pesar de la importancia de predecir la solubilidad, no es un asunto fácil. Una aproximación, utilizando simulaciones de "fuerza bruta", requiere tiempos de cálculo prolongados. Otras técnicas, mientras más rápido, no pueden predecir valores de solubilidad precisos. Esta semana en La Revista de Física Química , Los investigadores informan sobre un nuevo tipo de software que permite estimaciones convenientes de la solubilidad de prácticamente cualquier sustancia molecular en amplios rangos de temperatura y presión. El código hace uso de software de código abierto fácilmente disponible y se espera que sea ampliamente adoptado.
Daan Frenkel de la Universidad de Cambridge en el Reino Unido trabajó con sus colegas Lunna Li, también en Cambridge, y Tim Totton, de British Petroleum, para desarrollar el código.
"Tomamos la decisión consciente de utilizar software disponible gratuitamente porque queríamos que nuestro enfoque estuviera disponible para todos, "Dijo Frenkel." Hace mucho tiempo que falta una herramienta de propósito general para calcular las solubilidades. La metodología subyacente estaba ahí, pero nadie había creado realmente un programa de trabajo ".
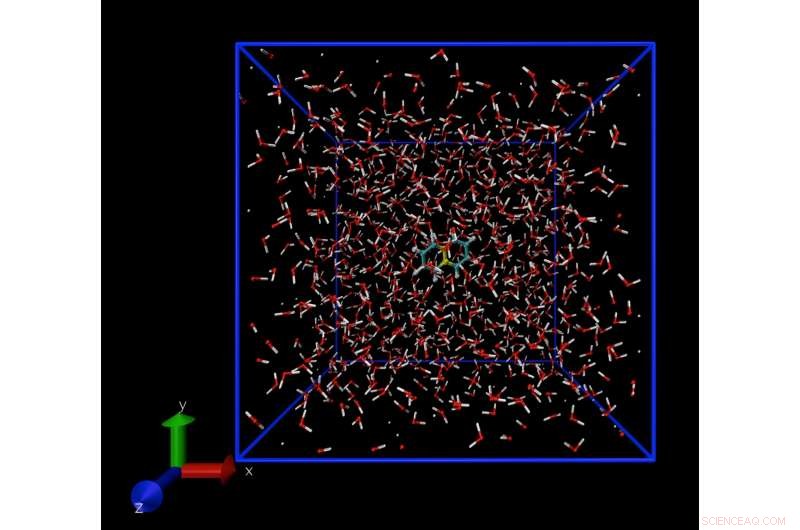
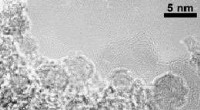
Una instantánea de una simulación de dinámica molecular que muestra una sola molécula de naftaleno, disuelto en agua. La técnica de simulación permite calcular la concentración de moléculas de naftaleno en agua en el límite de solubilidad. Crédito:Daan Frenkel, Universidad de Cambridge
El software desarrollado por este grupo utiliza expresiones termodinámicas estándar que se conocen desde mediados del siglo XIX, como la presión de vapor. El enfoque aprovecha el hecho de que cuando una fase sólida o líquida está en equilibrio, sus presiones de vapor son iguales. Cuando se calienta un líquido o un sólido, las moléculas escapan y forman vapor. Esta presión de vapor se puede calcular utilizando modelos informáticos.
Por ejemplo, un terrón de azúcar que se disuelve en agua:las moléculas de azúcar existen en un estado sólido (el terrón de azúcar cristalino) o completamente rodeadas por moléculas de agua una vez que se han disuelto. La cantidad de azúcar en cada una de las dos fases, sólido y solución, está determinada por la energía requerida para mover moléculas de azúcar entre esas fases. La solubilidad se puede calcular calculando la presión de vapor de las dos fases y equiparándolas.
Para modelar la fase sólida, los investigadores utilizaron un modelo denominado cristal de Einstein. En este modelo, Las moléculas de soluto que no interactúan se colocan en una red y se unen a un punto de la red con un resorte matemático. La presión de vapor del cristal se calcula calculando el trabajo necesario para apagar los resortes y activar las interacciones entre las moléculas atadas.
Para modelar una molécula de soluto disuelta, los investigadores utilizaron un potencial energético estándar para el disolvente en cuestión, que era agua en los ejemplos utilizados para probar su software, y calculó el trabajo en tres pasos. Primero, se crea una cavidad en el disolvente. Luego se inserta una molécula de soluto en la cavidad y, finalmente, la cavidad se reduce al tamaño de la molécula de soluto. Este procedimiento elimina una serie de errores y produce estimaciones precisas de la presión de vapor y, por lo tanto, la solubilidad.
En el informe de esta semana, los investigadores probaron su código en naftaleno disuelto en agua y predijeron una solubilidad que se compara bien con los valores experimentales. Las investigaciones futuras se centrarán en ampliar el software para que pueda manejar moléculas de soluto más grandes.