Ingenieros biomédicos de la Universidad de Duke han desarrollado una plataforma de inteligencia artificial que compara moléculas de forma autónoma y aprende de sus variaciones para anticipar diferencias de propiedades fundamentales para descubrir nuevos productos farmacéuticos. La plataforma proporciona a los investigadores una herramienta más precisa y eficiente para ayudar a diseñar terapias y otros productos químicos con propiedades útiles.
La investigación fue publicada el 27 de octubre en el Journal of Cheminformatics. .
Los algoritmos de aprendizaje automático se utilizan cada vez más para estudiar y predecir las propiedades biológicas, químicas y físicas de pequeñas moléculas utilizadas en el desarrollo de fármacos y otras tareas de diseño de materiales. Estas herramientas pueden ayudar a los investigadores a comprender las propiedades clave "ADMET" de una molécula:cómo se absorbe, distribuye, metaboliza, excreta y su toxicidad dentro del cuerpo. Al comprender estas diferentes propiedades, los investigadores pueden identificar moléculas para desarrollar nuevas terapias que sean más seguras y efectivas.
Si bien las plataformas de aprendizaje automático existentes permiten a los investigadores examinar una cantidad mucho mayor de moléculas de lo que sería posible fabricándolas todas físicamente en un laboratorio, solo pueden predecir las propiedades de una molécula a la vez, lo que limita su eficiencia general cuando se les asigna la tarea de identificar las compuesto más óptimo.
Si bien existen algunos otros enfoques computacionales para eliminar este paso adicional y comparar moléculas directamente, su alcance es limitado. Por ejemplo, métodos como la perturbación de energía libre son muy precisos pero computacionalmente tan complejos que sólo pueden evaluar un puñado de moléculas a la vez. Por otro lado, enfoques como los pares moleculares coincidentes son mucho más rápidos, pero solo pueden comparar moléculas muy similares, lo que limita su uso más amplio.
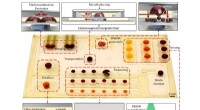
Para abordar esta cuestión, Reker y Zachary Fralish, Ph.D. Un estudiante del laboratorio de Reker desarrolló DeepDelta, un enfoque de aprendizaje profundo que puede comparar eficientemente dos moléculas simultáneamente y predecir las diferencias de propiedades entre ellas incluso si son muy diferentes.
"Al hacer que la red aprenda de una comparación uno a uno, le estás dando más puntos de datos que si estuviera aprendiendo de una molécula a la vez", dijo Reker. "La plataforma aprende sobre la estructura y propiedades de cada molécula individualmente, pero también aprende sobre las diferencias entre las dos y cómo esas diferencias influyen en las propiedades de la molécula".
El equipo probó la plataforma DeepDelta con dos modelos de última generación en el campo:Random Forest, un modelo clásico de aprendizaje automático ampliamente utilizado, y ChemProp, una red neuronal profunda en la que se basa DeepDelta. Cada sistema comparó dos estructuras moleculares conocidas y predijo 10 propiedades ADMET diferentes, incluida la forma en que las moléculas se eliminan de los riñones, sus respectivas vidas medias y qué tan bien pueden ser metabolizadas en el hígado.
DeepDelta demostró ser significativamente más eficaz y preciso a la hora de predecir y cuantificar las diferencias en las propiedades moleculares entre moléculas que las plataformas existentes.
"El entrenamiento sobre las diferencias moleculares permite que este método sea más preciso a la hora de decidir si una nueva sustancia química es mejor o peor que una actual", dijo Fralish. "Es como hacer una tarea que se parece más a una prueba. También ampliamos enormemente el tamaño de nuestros conjuntos de datos mediante el emparejamiento, esencialmente dando a nuestros modelos más tareas, lo que realmente ayuda a las redes neuronales ávidas de datos a aprender más".
El equipo ahora espera incorporar este modelo a su trabajo mientras diseñan nuevas terapias potenciales y optimizan los fármacos candidatos existentes.
"Con esta herramienta, podríamos analizar un medicamento que casi logró la aprobación de la FDA, pero tal vez tuvo problemas con la toxicidad hepática, por lo que no lo logró", dijo Fralish. "DeepDelta podría ayudar a identificar moléculas que tengan las mismas buenas propiedades pero sin toxicidad hepática. Esta herramienta abre muchas oportunidades al ayudarnos a decidir qué sustancia química tiene más posibilidades de hacer lo que queremos en el mundo real, ahorrando tiempo y dinero. "
Más información: Zachary Fralish et al, DeepDelta:predicción de mejoras ADMET de derivados moleculares con aprendizaje profundo, Journal of Cheminformatics (2023). DOI:10.1186/s13321-023-00769-x
Proporcionado por la Universidad de Duke