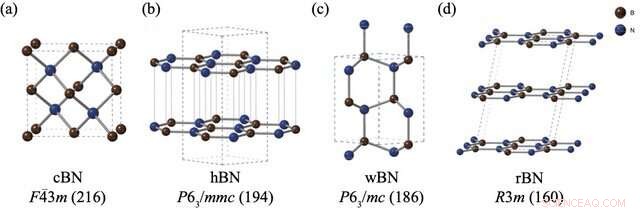
Las estructuras y grupos espaciales de (a) nitruro de boro zinc-blenda (cBN), (b) nitruro de boro hexagonal (hBN), (c) nitruro de boro wurtzita (wBN) y (d) nitruro de boro romboédrico (rBN). Los átomos de boro y nitrógeno se representan en marrón y azul, respectivamente. Crédito:Kousuke Nakano de JAIST.
El nitruro de boro (BN) es un material versátil con aplicaciones en una variedad de campos científicos y de ingeniería. Esto se debe en gran parte a una propiedad interesante de BN llamada "polimorfismo", caracterizada por la capacidad de cristalizar en más de un tipo de estructura. Esto generalmente ocurre como una respuesta a los cambios de temperatura, presión o ambos. Además, las diferentes estructuras, denominadas "polimorfos", difieren notablemente en sus propiedades físicas a pesar de tener la misma fórmula química. Como resultado, los polimorfos juegan un papel importante en el diseño de materiales, y el conocimiento de cómo favorecer selectivamente la formación del polimorfo deseado es crucial en este sentido.
Sin embargo, los polimorfos de BN plantean un problema particular. A pesar de realizar varios experimentos para evaluar las estabilidades relativas de los polimorfos de BN, no ha surgido un consenso sobre este tema. Si bien los métodos computacionales son a menudo el enfoque de acceso para estos problemas, los polimorfos BN han planteado serios desafíos a las técnicas de computación estándar debido a las débiles "interacciones de van der Waals (vdW)" entre sus capas, que no se tienen en cuenta en estos cálculos. Además, los cuatro polimorfos estables de BN, a saber, romboédrico (rBN), hexagonal (hBN), wurtzita (wBN) y zinc-blenda (cBN), se manifiestan dentro de un rango de energía estrecho, lo que hace que la captura de pequeñas diferencias de energía junto con interacciones vdW aún más desafiante.
Un equipo de investigación internacional dirigido por el profesor asistente Kousuke Nakano del Instituto Avanzado de Ciencia y Tecnología de Japón (JAIST) ahora ha proporcionado evidencia para resolver el debate. En su estudio, abordaron el problema con un marco de cálculo de principios básicos de última generación, a saber, simulaciones Monte Carlo de difusión de nodo fijo (FNDMC). FNDMC representa un paso en el popular método cuántico de simulación Monte Carlo, en el que una "función de onda" cuántica de muchos cuerpos parametrizada se optimiza primero para alcanzar el estado fundamental y luego se suministra a FNDMC.
Además, el equipo también calculó la energía de Gibbs (el trabajo útil que se puede obtener de un sistema a presión y temperatura constantes) de los polimorfos de BN para diferentes temperaturas y presiones utilizando la teoría funcional de la densidad (DFT) y cálculos de fonones. Este artículo estuvo disponible en línea el 24 de marzo de 2022 y se publicó en The Journal of Physical Chemistry C .
Según los resultados de la FNDMC, hBN fue la estructura más estable, seguida de rBN, cBN y wBN. Estos resultados fueron consistentes tanto a 0 K como a 300 K (temperatura ambiente). Sin embargo, las estimaciones de DFT arrojaron resultados contradictorios para dos aproximaciones diferentes. El Dr. Nakano explica estos hallazgos contradictorios:"Nuestros resultados revelan que la estimación de las estabilidades relativas está muy influenciada por el funcional de correlación de intercambio o la aproximación utilizada en el cálculo de DFT. Como resultado, no se puede llegar a una conclusión cuantitativa utilizando los hallazgos de DFT, y se requiere un enfoque más preciso, como FNDMC".
En particular, los resultados de FNDMC coincidieron con los generados por otros métodos de cálculo refinados, como el "clúster acoplado", lo que sugiere que FNDMC es una herramienta eficaz para lidiar con polimorfos, especialmente aquellos gobernados por fuerzas vdW. El equipo también demostró que puede proporcionar otra información importante, como energías de referencia confiables, cuando los datos experimentales no están disponibles.
El Dr. Nakano está entusiasmado con las perspectivas futuras del método en el área de la ciencia de los materiales. "Nuestro estudio demuestra la capacidad de FNDMC para detectar pequeños cambios de energía que involucran fuerzas vdW, lo que estimulará el uso de este método para otros materiales de van der Waals", dice. "Además, las simulaciones moleculares basadas en este método preciso y confiable podrían potenciar los diseños de materiales, lo que permitiría el desarrollo de medicamentos y catalizadores". Aumento de la precisión de los cálculos de la fuerza atómica con la transformación de coordenadas de deformación espacial