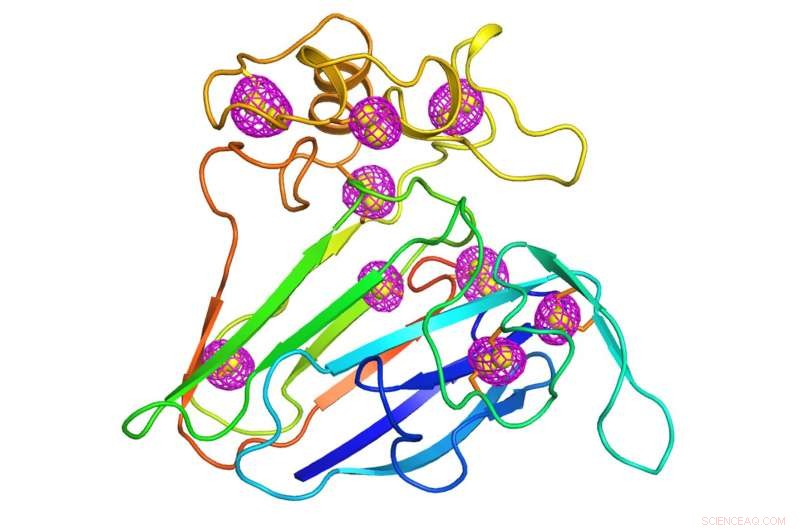
Una caricatura que representa la estructura de una proteína vegetal bien estudiada que sirvió como caso de prueba para la técnica de microcristalografía recientemente desarrollada. Los patrones de malla magenta que rodean los átomos de azufre intrínsecos a la proteína (esferas amarillas) indican las señales anómalas que se extrajeron mediante difracción de rayos X de baja energía de miles de cristales que miden menos de 10 millonésimas de metro. del tamaño de una bacteria. Crédito:Laboratorio Nacional Brookhaven
El uso de rayos X para revelar las estructuras tridimensionales a escala atómica de las proteínas ha llevado a innumerables avances en la comprensión de cómo funcionan estas moléculas en las bacterias. virus plantas y humanos, y ha guiado el desarrollo de medicamentos de precisión para combatir enfermedades como el cáncer y el SIDA. Pero muchas proteínas no se pueden convertir en cristales lo suficientemente grandes como para descifrar sus arreglos atómicos. Para afrontar este desafío, Los científicos del Laboratorio Nacional Brookhaven del Departamento de Energía de EE. UU. (DOE) y sus colegas de la Universidad de Columbia han desarrollado un nuevo enfoque para resolver estructuras de proteínas a partir de cristales diminutos.
El método se basa en un manejo de muestras único, extracción de señal, y enfoques de ensamblaje de datos, y una línea de luz capaz de enfocar rayos X intensos en la Fuente de luz sincrotrón nacional II de Brookhaven (NSLS-II), una instalación para usuarios de la Oficina de Ciencias del DOE, hasta un punto de una millonésima de metro, aproximadamente una quincuagésima parte del ancho de un cabello humano.
"Nuestra técnica realmente abre la puerta para lidiar con microcristales que antes eran inaccesibles, incluyendo receptores de superficie celular difíciles de cristalizar y otras proteínas de membrana, proteínas flexibles, y muchas proteínas humanas complejas, "dijo el científico de Brookhaven Lab, Qun Liu, el autor correspondiente del estudio, que fue publicado el 3 de mayo, 2019, en IUCrJ , una revista de la Unión Internacional de Cristalografía.
Descifrando estructuras proteicas
La cristalografía de proteínas ha sido un método dominante para resolver estructuras de proteínas desde 1958, mejorando con el tiempo a medida que las fuentes de rayos X se han vuelto más potentes, permitiendo determinaciones de estructura más precisas. Para determinar la estructura de una proteína, Los científicos miden cómo los rayos X como los generados en NSLS-II difractan, o rebotar, los átomos en una red cristalina ordenada que consta de muchas copias de la misma molécula de proteína, todas dispuestas de la misma manera. El patrón de difracción transmite información sobre dónde se encuentran los átomos. Pero no es suficiente.
"Solo las amplitudes de las 'ondas' de rayos X difractadas se registran en el detector, pero no sus fases (el tiempo entre ondas), "dijo Liu." Ambos son necesarios para reconstruir una estructura 3-D. Este es el llamado problema de la fase cristalográfica ".
Los cristalógrafos han resuelto este problema recopilando datos de fase de un tipo diferente de dispersión, conocido como dispersión anómala. La dispersión anómala ocurre cuando los átomos más pesados que los componentes principales de carbono de una proteína, hidrógeno, y el nitrógeno absorben y reemiten algunos de los rayos X. Esto sucede cuando la energía de los rayos X está cerca de la energía que a esos átomos pesados les gusta absorber. Los científicos a veces insertan artificialmente átomos pesados como el selenio o el platino en la proteína para este propósito. Pero los átomos de azufre, que aparecen de forma natural a lo largo de las moléculas de proteína, también puede producir tales señales, aunque más débil. Aunque estas señales anómalas son débiles, un gran cristal por lo general tiene suficientes copias de la proteína con suficientes átomos de azufre para que sean medibles. Eso les da a los científicos la información de fase necesaria para identificar la ubicación de los átomos de azufre y traducir los patrones de difracción en una estructura tridimensional completa.
"Una vez que conozca las posiciones del azufre, puede calcular las fases para los otros átomos de proteína porque la relación entre el azufre y los otros átomos es fija, "dijo Liu.
Pero pequeños cristales por definición, no tiene tantas copias de la proteína de interés. Entonces, en lugar de buscar información de difracción y fase a partir de copias repetidas de una proteína en un solo cristal grande, el equipo de Brookhaven / Columbia desarrolló una forma de tomar medidas de muchos cristales diminutos, y luego reunir los datos colectivos.
Pequeños cristales grandes resultados
Para manejar los diminutos cristales el equipo desarrolló cuadrículas de muestras con patrones de pozos de tamaño micro. Después de verter el disolvente que contiene los microcristales sobre estas rejillas bien montadas, los científicos eliminaron el solvente y congelaron los cristales que estaban atrapados en las rejillas.
"Todavía tenemos un desafío, aunque, porque no podemos ver dónde están los cristales diminutos en nuestra cuadrícula, "dijo Liu." Para averiguarlo, Utilizamos microdifracción en la línea de luz de Cristalografía Macromolecular Microfocalizadora Frontier (FMX) de NSLS-II para estudiar toda la cuadrícula. Escaneando línea por línea, podemos encontrar dónde están escondidos esos cristales ".
Como Martin Fuchs, el científico líder en línea de luz en FMX, explicado, "La línea de luz FMX puede enfocar toda la intensidad del haz de rayos X hasta un tamaño de un micrón, o millonésima de metro. Podemos controlar con precisión el tamaño del haz para que coincida con el tamaño de los cristales, cinco micrones en el caso del experimento actual. Estas capacidades son cruciales para obtener la mejor señal, " él dijo.
Wuxian Shi, otro científico de línea de luz FMX, señaló que "los datos recopilados en la encuesta de cuadrícula contienen información sobre la ubicación de los cristales. Además, También podemos ver qué tan bien difracta cada cristal, lo que nos permite elegir solo los mejores cristales para la recopilación de datos ".
Luego, los científicos pudieron maniobrar el soporte de la muestra para colocar cada microcristal de interés mapeado nuevamente en el centro del haz de rayos X de precisión para la recolección de datos.
Utilizaron la energía más baja disponible en la línea de luz, sintonizada para aproximarse lo más posible a la energía de absorción de los átomos de azufre, y recopilaron datos de dispersión anómalos.
"La mayoría de las líneas de luz cristalográficas no pudieron alcanzar el borde de absorción de azufre para optimizar las señales anómalas, ", dijo el coautor Wayne Hendrickson de la Universidad de Columbia." Afortunadamente, NSLS-II es una fuente de luz de sincrotrón líder en el mundo que proporciona rayos X brillantes que cubren un amplio espectro de energía de rayos X. Y aunque nuestro nivel de energía estaba ligeramente por encima de la energía de absorción ideal para el azufre, generó las señales anómalas que necesitábamos ".
Pero los científicos aún tenían trabajo por hacer para extraer esas señales importantes y reunir los datos de muchos cristales diminutos.
"De hecho, estamos obteniendo miles de datos, "dijo Liu." Usamos alrededor de 1400 microcristales, cada uno con su propio conjunto de datos. Tenemos que juntar todos los datos de esos microcristales ".
They also had to weed out data from crystals that were damaged by the intense x-rays or had slight variations in atomic arrangements.
"A single microcrystal does not diffract x-rays sufficiently for structure solution prior to being damaged by the x-rays, " said Sean McSweeney, deputy photon division director and program manager of the Structural Biology Program at NSLS-II. "This is particularly true with crystals of only a few microns, the size of about a bacterial cell. We needed a way to account for that damage and crystal structure variability so it wouldn't skew our results."
They accomplished these goals with a sophisticated multi-step workflow process that sifted through the data, discarded outliers that might have been caused by radiation damage or incompatible crystals, and ultimately extracted the anomalous scattering signals.
"This is a critical step, " said Liu. "We developed a computing procedure to assure that only compatible data were merged in a way to align the individual microcrystals from diffraction patterns. That gave us the required signal-to-noise ratios for structure determination."
Applying the technique
This technique can be used to determine the structure of any protein that has proven hard to crystallize to a large size. These include cell-surface receptors that allow cells of advanced lifeforms such as animals and plants to sense and respond to the environment around them by releasing hormones, transmitting nerve signals, or secreting compounds associated with cell growth and immunity.
"To adapt to the environment through evolution, these proteins are malleable and have lots of non-uniform modifications, " said Liu. "It's hard to get a lot of repeat copies in a crystal because they don't pack well."
Inhumanos, receptors are common targets for drugs, so having knowledge of their varied structures could help guide the development of new, more targeted pharmaceuticals.
But the technique is not restricted to just small crystals.
"The method we developed can handle small protein crystals, but it can also be used for any size protein crystals, any time you need to combine data from more than one sample, "Dijo Liu.