Crédito:Universidad de California, San Francisco
El descubrimiento de fármacos puede traer a la mente imágenes de batas de laboratorio y pipetas blancas, pero cuando Henry Lin, Doctor, recientemente se propuso encontrar un mejor opioide con menos efectos secundarios, su primer paso fue encender las computadoras.
Usando un programa llamado DOCK, Cargó una estructura cristalina del receptor opioide que se encuentra en el cerebro y accedió a una biblioteca virtual de 3 millones de compuestos que podrían unirse a un "bolsillo" químico en el receptor. La mayoría de los medicamentos, desde los antibióticos hasta los antidepresivos, funcionan uniéndose a sitios específicos de las proteínas, pero para ser efectivo deben encajar a la perfección.
El programa hizo girar cada compuesto, consideró la flexibilidad de sus diversos apéndices, y después de probar un promedio de 1.3 millones de configuraciones por compuesto, las clasificó por su potencial de unión. El proceso, ejecutándose en computadoras conectadas a procesadores potentes, tomó alrededor de dos semanas.
Un estudiante de posgrado en ese momento, Lin trabajó con su asesor Brian Shoichet, Doctor, profesor de química farmacéutica en la Facultad de Farmacia de UC San Francisco, y Aashish Manglik, Doctor, de la Universidad de Stanford para peinar los 2 primeros, 500 compuestos para factores adicionales y 23 seleccionados para pruebas experimentales en células vivas:batas de laboratorio y pipetas.
Cada vez más, los investigadores están recurriendo a experimentos virtuales para los pasos iniciales del desarrollo de fármacos. Con computadoras cada vez más rápidas, la fase inicial y en gran parte de prueba y error del desarrollo de un fármaco puede reducirse a cuestión de días, y con bibliotecas en línea de compuestos en constante expansión, las pruebas de detección de drogas pueden abarcar, literalmente, toda la química conocida en el mundo.
Fortalezas y limitaciones
Los investigadores son cautelosos sobre el potencial del descubrimiento de fármacos computacionales (solo una pequeña fracción de compuestos prometedores realmente funcionan cuando se prueban en la vida real), pero dicen que uno de sus puntos fuertes es revelar compuestos completamente nuevos como candidatos a fármacos.
Shoichet se especializa en un método computacional popular conocido como acoplamiento molecular. "Donde encaja el acoplamiento es en la investigación de descubrimiento temprano, en la búsqueda de nuevas salidas, " él dijo.
La búsqueda de su equipo del nuevo opioide ilustra tanto las fortalezas como las limitaciones del descubrimiento de fármacos computacionales.
De hecho, los candidatos a opioides iniciales identificados mediante acoplamiento molecular se desempeñaron solo de manera modesta en las pruebas experimentales. "Todavía, la actividad que tenían era muy reproducible y las moléculas muy novedosas, presagiando una nueva biología, "dijo Shoichet.
El equipo acopló otra ronda de compuestos con estructuras similares y probó a los máximos anotadores. Con colaboradores en la Universidad de Carolina del Norte, Chapel Hill y la Universidad Friedrich Alexander en Alemania, identificaron el compuesto más potente y optimizaron su farmacología con elaboración sintética guiada por computadora.
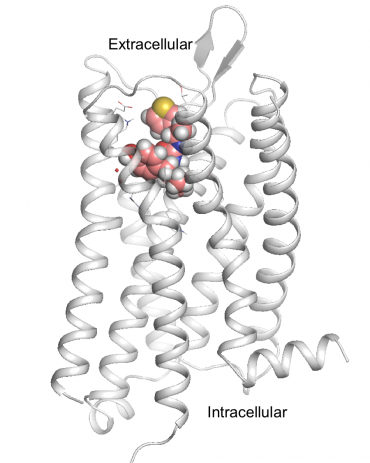
PZM21, el nuevo, candidato a fármaco opioide más seguro, se muestra acoplado al receptor de morfina del cerebro, el receptor mu-opioide. Crédito:Anat Levit
Ese compuesto ganador llamado PZM21, es químicamente diferente a cualquiera de los que se utilizan actualmente y es posible que no se haya encontrado mediante métodos más tradicionales. Es un compuesto diseñado completamente computacionalmente que es más potente que la morfina. En ratones, bloqueaba eficazmente el dolor sin los efectos secundarios habituales de la supresión respiratoria y el estreñimiento e incluso parecía ser menos adictivo.
El acoplamiento no es una bala de plata pero se ha convertido en un poderoso punto de partida durante mucho tiempo, proceso interdisciplinario de desarrollo de fármacos. Entre sus principales contribuciones se encuentran los inhibidores de proteasa que han contribuido a que el VIH sea una enfermedad tratable. Los investigadores también están utilizando el acoplamiento para detectar candidatos a fármacos para el tratamiento del cáncer de mama, hepatitis C, hipertensión, Estafilococo, el virus del SARS y la influenza.
Tecnología pionera en UCSF
El acoplamiento molecular fue iniciado hace tres décadas por un joven químico físico de UCSF llamado Tack Kuntz, Doctor, ahora profesor emérito de la Facultad de Farmacia. Cuando Kuntz llegó al campus a principios de la década de 1970, todavía prevalecía el enfoque tradicional para el descubrimiento de fármacos.
Como lo describió Kuntz, el proceso se basaba en el azar y en muy poca teoría:"Uno sale y encuentra nuevos compuestos naturales y los trae de vuelta para probarlos en un laboratorio. Simplemente mezcle los productos químicos con un organismo y vea qué sucede".
Los químicos farmacéuticos apenas pensaron en los detalles moleculares de cómo las drogas interactúan con el cuerpo. Muchas drogas, incluidos los primeros antibióticos, había sido descubierto por casualidad, pero Kuntz, habiendo visto la nueva comprensión molecular arrasando el campo de la biología, sintió que era hora de una actualización similar en farmacología.
"La visión de la biología basada en objetivos, que puede comprender la biología a través de proteínas independientes y productos genéticos, ya se había hecho cargo, pero la farmacología estaba una década atrás, "dijo Shoichet, quien era un estudiante de posgrado en el laboratorio de Kuntz en la década de 1980.
Kuntz y sus colegas comenzaron a trabajar hacia un enfoque más racional del diseño de fármacos en el que intentaron identificar compuestos que pudieran encajar receptores específicos en proteínas. como encontrar la pieza que falta en un rompecabezas. En 1982, publicaron un artículo que describe el primer programa de acoplamiento molecular que podría "explorar alineaciones geométricamente factibles de ligandos y receptores de estructura conocida".
Kuntz envió 10, 000 copias de ese primer programa de acoplamiento a investigadores de todo el país. Pronto, otros investigadores estaban desarrollando programas computacionales similares y la emoción se extendió rápidamente fuera de la academia. En la década de 1990, todas las compañías farmacéuticas importantes habían abierto una unidad de descubrimiento de fármacos computacional.
Ponerse al día con una idea
A pesar del entusiasmo inicial, sin embargo, El descubrimiento de fármacos computacionales no condujo a resultados rápidos. La idea de Kuntz había llegado antes de tiempo. Se necesitarían décadas de avances incrementales en biología molecular, tecnología de imágenes y computación, antes de que el descubrimiento de fármacos computacionales pudiera comenzar a cumplir su promesa.

Tack Kuntz, Doctor, y sus colegas en 1982 publicaron un artículo que describe el primer programa de acoplamiento molecular que podría "explorar alineaciones geométricamente factibles de ligandos y receptores de estructura conocida". Crédito:Universidad de California, San Francisco
Una limitación importante en la década de 1990 fue la falta de estructuras proteicas conocidas. Sin estos había pocos objetivos para los que encontrar drogas. En las décadas posteriores, La cristalografía de rayos X y la resonancia magnética nuclear han revelado miles de estructuras de proteínas de posibles dianas de fármacos.
El descubrimiento del nuevo candidato a opioide, por ejemplo, fue posible solo debido a las estructuras cristalinas recientemente determinadas de los receptores acoplados a proteína G, una familia de proteínas que incluye el receptor opioide.
Las bibliotecas virtuales de compuestos también han crecido exponencialmente. En 1991, una base de datos puede contener 55, 000 compuestos; ahora contienen decenas de millones. "El alcance de la química que estamos muestreando ha aumentado aproximadamente al mismo ritmo que la Ley de Moore, "Hay un hambre insaciable de más y más moléculas", dijo Shoichet.
Los programas de acoplamiento de hoy en día pueden modelar de manera realista las interacciones a nivel atómico entre un fármaco y su objetivo, pero algunos detalles delicados, como cómo cambian las fuerzas atómicas cuando una molécula de fármaco desplaza el agua en el sitio de unión, siguen siendo desafíos continuos en el campo.
Promesas y pruebas
El acoplamiento molecular no es la única forma de diseño de fármacos por computadora. En el Instituto de Ciencias de la Salud Computacional de la UCSF (ICHS), decenas de investigadores están explorando innumerables métodos computacionales para avanzar en la investigación médica.
Michael Keizer, Doctor, miembro del ICHS y profesor asistente en el Instituto de Enfermedades Neurodegenerativas, está estudiando fármacos que atacan a muchos objetivos moleculares a la vez, como si tocara un acorde en lugar de una sola nota. Durante mucho tiempo se entendió que esta acción de objetivos múltiples era la causa de efectos secundarios no deseados, pero también puede dirigirse al tratamiento de enfermedades complejas.
Solo a principios de la década de 2000 los investigadores llegaron a reconocer que muchos fármacos existentes funcionan a través de más de un objetivo:los antipsicóticos, por ejemplo, que afectan tanto a los receptores de serotonina como a los de dopamina. Ahora están diseñando medicamentos intencionalmente para hacerlo.
"Para algunas enfermedades que aún no tienen tratamiento, tal vez sea porque no hay una sola proteína que deba activar o desactivar; ¿Qué pasa si la droga necesita alcanzar múltiples objetivos en su lugar? ", dijo Keizer, quien era un estudiante de posgrado de Shoichet.
En su laboratorio, Keizer utiliza métodos computacionales para identificar patrones químicos entre fármacos que se unen al mismo conjunto de objetivos y encontrar nuevos compuestos que tengan una farmacología coincidente. Este enfoque computacional puede reconocer similitudes entre compuestos que los análisis más convencionales pasarían por alto. Keizer ahora está mirando hacia la tecnología de inteligencia artificial, conocido como aprendizaje profundo, para un mejor reconocimiento de patrones.
Incluso cuando los métodos computacionales despegan, su prueba todavía está en el mundo real:en las celdas, modelos animales, y finalmente en la clínica. "Durante un tiempo fue común publicar artículos con predicciones sobre las actividades de una molécula pequeña, pero ninguna prueba real de estas predicciones, porque los experimentos para hacerlo eran costosos, difícil o esotérico, "dijo Keizer.
A medida que se ha vuelto clara la necesidad de colaboración, la asociación entre la predicción computacional y los experimentos de laboratorio húmedo se ha fortalecido notablemente en la última década, dijo Keizer. "Después de todo, ¿Cómo puede mejorar sus predicciones si no está seguro de cuáles son las incorrectas? "