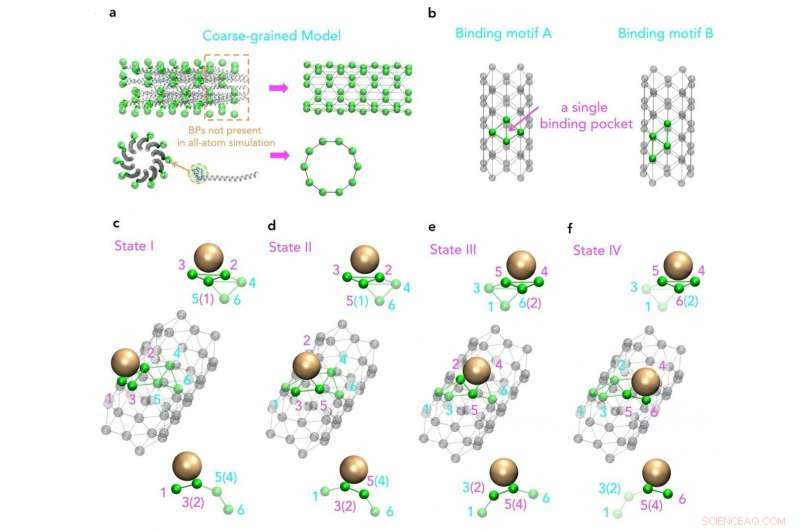
Vista molecular de un modelo de grano grueso basado en la estructura original de las principales proteínas de la cubierta M13 Crédito:SUTD
Las simulaciones atomísticas son una herramienta poderosa para estudiar el movimiento y las interacciones de átomos y moléculas. En muchos procesos biológicos, efectos a gran escala, por ejemplo, El ensamblaje de grandes virus en nanopartículas es importante. Los procesos de ensamblaje de estos grandes virus son de fundamental importancia para el diseño de muchos dispositivos y terapias dirigidas a proteínas virales. Sin embargo, la escala de tiempo y duración de estos procesos de ensamblaje suele ser demasiado grande para simulaciones con resolución molecular.
Es más, a pesar de que un aumento en la potencia de cálculo permite simulaciones más complejas y más largas, estructuras de virus, como M13, todavía están fuera de su alcance. Es por eso que un grupo de investigación de la Universidad de Tecnología y Diseño de Singapur (SUTD) y el Instituto de Tecnología de Massachusetts (MIT) ha desarrollado un procedimiento que vincula los procesos de ensamblaje a gran escala con las simulaciones moleculares. Profesor asistente Desmond Loke de Ciencias de SUTD, El grupo de Matemáticas y Tecnología dijo:"Para la simulación de M13, comenzamos con diferentes conjuntos de campos de fuerza. Se eligieron campos de fuerza adecuados y se utilizaron como entradas para simulaciones de dinámica de moléculas con el modelo de grano grueso diseñado para capturar el patrón clave del proceso de ensamblaje ".
"Si bien sabemos que la fabricación basada en M13 puede estar impulsada fundamentalmente por interacciones nanopartícula-péptido, que también puede ser un principio clave detrás de la bioingeniería de tipo M13, tenemos poco conocimiento de cómo los patrones repetidos de péptidos de extremo corto en una superficie M13 están realmente involucrados en estas interacciones. Para estudiar esto, idealmente tenemos que incluir una estructura completa de la proteína de la cubierta viral, que es una tarea difícil para las simulaciones actuales de dinámica molecular de última generación, "agrega la Dra. Lunna Li, primer autor del artículo.
El procedimiento permite a los usuarios agregar diferentes tipos de nanopartículas a una solución, a un nivel realista. Inspirado por este procedimiento, El profesor asistente Loke y sus colegas pudieron simular un virus a gran escala con nanopartículas y dentro de una solución durante cincuenta nanosegundos.
El Dr. Li dijo:"La estructura y la solución del virus contienen alrededor de 700, 000 átomos en total ". Teniendo en cuenta la forma y el tamaño de las características, la complejidad de esta simulación puede ser mayor que cualquier simulación realizada anteriormente.
"Una simulación realizada en microsegundos habría sido posible si se hubiera utilizado un modelo M13 más pequeño, pero puede valer la pena reducir el tiempo para observar realmente cómo la estructura completa puede influir en el ensamblaje entre el M13 y las nanopartículas, "explicó el profesor asistente Loke.