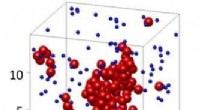
Una formación de estructuras en forma de cadena en una superficie de cobre a partir del autoensamblaje molecular, según lo predicho por un nuevo método computacional. Estas estructuras en forma de cadena pueden funcionar como pequeños alambres con diámetros de 1/100, Milésima parte de un mechón de cabello para futuros dispositivos eléctricos. Crédito:Universidad de Kioto iCeMS
Daniel Packwood, Profesor asociado junior en el Instituto de Ciencias Integradas del Material Celular de la Universidad de Kioto (iCeMS), está mejorando los métodos para construir diminutos "nanomateriales" utilizando un enfoque "de abajo hacia arriba" llamado "autoensamblaje molecular". Usando este método, las moléculas se eligen de acuerdo con su capacidad para interactuar espontáneamente y combinarse para formar formas con funciones específicas. En el futuro, este método se puede utilizar para producir alambres diminutos con diámetros de 1/100, 000 la de un mechón de cabello, o pequeños circuitos eléctricos que pueden caber en la punta de una aguja.
El autoensamblaje molecular es un proceso espontáneo que no puede controlarse directamente con equipos de laboratorio, por lo que debe controlarse indirectamente. Esto se hace eligiendo cuidadosamente la dirección de las interacciones intermoleculares, conocido como "control químico", y elegir cuidadosamente la temperatura a la que ocurren estas interacciones, conocido como "control entrópico".
Los investigadores saben que cuando el control entrópico es muy débil, por ejemplo, las moléculas están bajo control químico y se ensamblan en la dirección de los sitios libres disponibles para la interacción molécula a molécula. Por otra parte, el autoensamblaje no ocurre cuando el control entrópico es mucho más fuerte que el control químico, y las moléculas permanecen dispersas al azar.
Hasta ahora, No ha sido posible para los investigadores adivinar qué tipos de estructuras resultarán del autoensamblaje molecular cuando el control entrópico no es ni débil ni fuerte en comparación con el control químico.
Packwood se asoció con colegas en Japón y los EE. UU. Para desarrollar un método computacional que les permite simular el autoensamblaje molecular en superficies metálicas mientras separa los efectos de los controles químicos y entrópicos.
Este nuevo método computacional hace uso de inteligencia artificial para simular cómo se comportan las moléculas cuando se colocan sobre una superficie metálica. Específicamente, Se utiliza una técnica de "aprendizaje automático" para analizar una base de datos de interacciones intermoleculares. Esta técnica de aprendizaje automático construye un modelo que codifica la información contenida en la base de datos, ya su vez, este modelo puede predecir el resultado del proceso de autoensamblaje molecular con alta precisión.
El equipo utilizó este método para estudiar el autoensamblaje de tres moléculas de hidrocarburos diferentes, cuyas estructuras varían en la fuerza de la dirección de sus interacciones intermoleculares. En otras palabras, variaron la fuerza del control químico cambiando la molécula en estudio.
Mientras que un control químico más fuerte hizo que las moléculas se ensamblaran en estructuras en forma de cadena, Se encontró que los efectos de controles entrópicos más fuertes eran más contradictorios. Por ejemplo, encontraron que fortalecer el control entrópico podría transformar grandes, estructuras desordenadas en varias pequeñas, ordenado, estructuras en forma de cadena. También demostraron que la formación de estructuras desordenadas resulta de un control químico débil más que de un control entrópico fuerte.
Estas predicciones, que se verificaron mediante comparaciones con imágenes microscópicas de alta resolución de moléculas reales en superficies metálicas, puede conducir a un control, Fabricación a gran escala de diminutos cables eléctricos y otros nanomateriales para dispositivos futuros. Los dispositivos fabricados con nanomateriales serían significativamente más pequeños y más baratos que los dispositivos electrónicos existentes. y tendría una vida útil muy larga de la batería debido al bajo consumo de energía.
"Mediante el desarrollo continuo de nuestro código y nuestra teoría, esperamos obtener reglas cada vez más detalladas para controlar el autoensamblaje molecular y ayudar al proceso de fabricación de nanomateriales de abajo hacia arriba, "concluyen los investigadores en su estudio publicado en la revista Comunicaciones de la naturaleza .