Al igual que los humanos que las crearon, a las computadoras les resulta difícil la física, pero aún más difícil la mecánica cuántica. Pero una nueva técnica creada por tres científicos de la Universidad de Chicago permite a las computadoras simular ciertos efectos desafiantes de la mecánica cuántica en materiales electrónicos complejos con mucho menos esfuerzo.
Al hacer que estas simulaciones sean más precisas y eficientes, los científicos esperan que la técnica pueda ayudar a descubrir nuevas moléculas y materiales, como nuevos tipos de células solares o computadoras cuánticas.
"Este avance tiene un inmenso potencial para mejorar nuestra comprensión de los fenómenos moleculares, con importantes implicaciones para la química, la ciencia de los materiales y campos relacionados", dijo el científico Daniel Gibney, Ph.D. de la Universidad de Chicago. estudiante de química y primer autor del artículo, publicado el 14 de diciembre en Physical Review Letters .
Una hoja o un panel solar parecen suaves y simples desde el exterior, pero si te acercas al nivel molecular verás una danza tremendamente complicada de electrones y moléculas.
Para lograr nuevos avances en sostenibilidad, manufactura, agricultura y muchos otros campos, los científicos modelan el comportamiento de estas interacciones químicas y moleculares. Esto ayuda a revelar nuevas posibilidades de diseño para el futuro, desde nuevas formas de secuestrar dióxido de carbono hasta nuevos tipos de bits cuánticos.
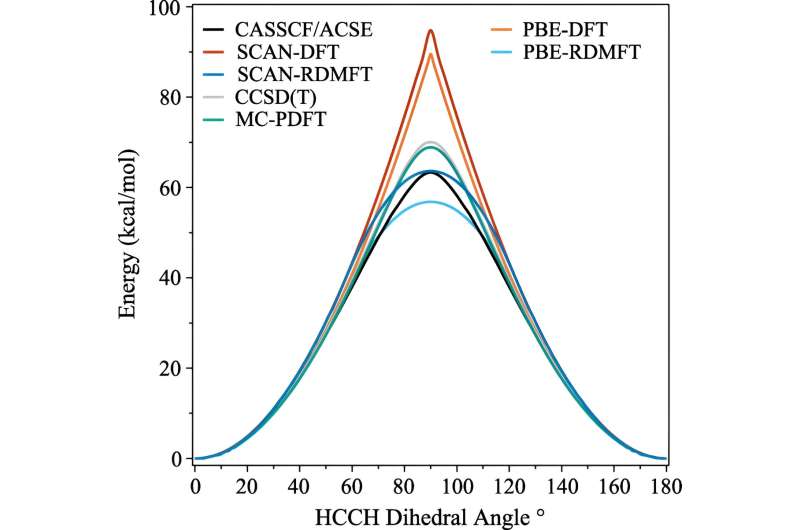
Se han logrado muchos avances en las últimas décadas, pero una de las áreas que sigue siendo difícil de simular es cuando las moléculas comienzan a mostrar comportamientos mecánicos cuánticos complejos que los científicos llaman correlación fuerte.
El problema es que una vez que los electrones empiezan a hacer alarde de sus efectos más mecánicos cuánticos (como "entrelazarse"), los cálculos necesitan instantáneamente mucha más potencia de cálculo. Incluso las supercomputadoras luchan por manejar las implicaciones.
Uno de los cálculos comúnmente utilizados se llama teoría funcional de la densidad. "Esta es básicamente la técnica más omnipresente para predecir la estructura electrónica, pero es esencialmente una aproximación en la que todos los electrones se tratan como una función de un electrón", explicó David Mazziotti, profesor de química y autor principal del estudio.
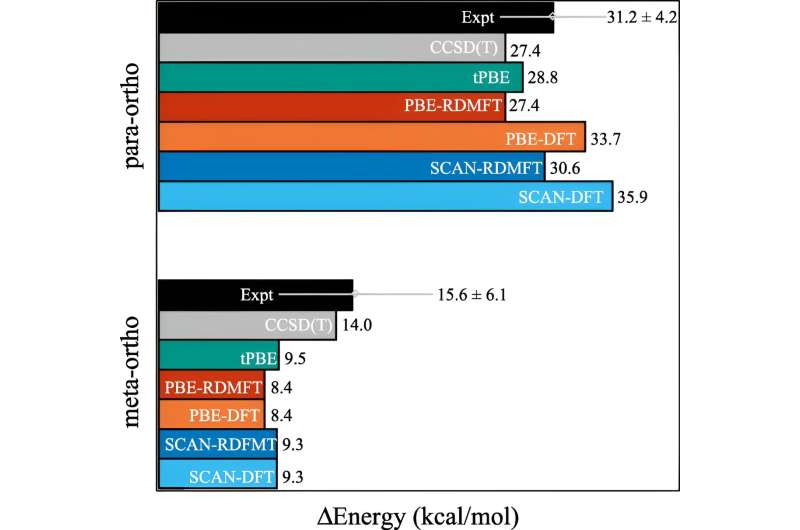
Para muchos cálculos, una aproximación es suficiente. Pero comienza a descomponerse a medida que el comportamiento de los electrones se vuelve más correlacionado, como sucede cuando la mecánica cuántica comienza a entrar en juego. En mecánica cuántica, estos electrones pueden estar en múltiples lugares u orbitales simultáneamente. Esto obstaculiza no sólo el cerebro humano, sino también la teoría funcional de la densidad.
"Y este es un problema importante, porque muchos de los temas que nos preocupan en el siglo XXI, como las nuevas moléculas y materiales para la energía renovable y la sostenibilidad, requieren que explotemos la naturaleza cuántica de los materiales", afirmó Mazziotti.
Mazziotti, Gibney y el tercer autor, Jan-Niklas Boyn, descubrieron que podían agregar una corrección universal a la teoría funcional de la densidad que permite que los electrones se enreden entre múltiples orbitales a la vez.
"Esto permite que los orbitales en el cálculo no sólo estén completamente llenos o completamente vacíos, sino también en cualquier punto intermedio", dijo Mazziotti. "Llegamos a una imagen de un solo electrón que aún es capaz de capturar el comportamiento que surge de los efectos correlacionados de electrones de muchos cuerpos."
Como beneficio adicional, dijeron los científicos, el código se puede agregar a los algoritmos existentes sin tener que reescribir ese código. "Básicamente, la corrección se activa cuando es necesario, pero de lo contrario no interfiere con el resto del código", dijo Gibney.
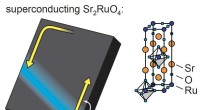
También es universal, ya que puede agregarse a códigos que simulen muchos tipos de comportamiento electrónico, ya sean paneles solares fotovoltaicos o secuestro de carbono o materiales superconductores, o incluso biología.
Por ejemplo, explicó Boyn, una aplicación podría ser la comprensión de la química que tiene lugar utilizando enzimas que contienen átomos metálicos, conocidas como metaloenzimas.
"Existe una gran cantidad de metaloenzimas responsables de gran parte de la química en las células, por ejemplo, pero han sido notoriamente difíciles de describir con los modelos actuales", dijo. "Esta teoría podría permitirnos en un futuro próximo abordar esta química de una manera que hoy es imposible."
Más información: Daniel Gibney et al, Generalización universal de la teoría funcional de la densidad para la correlación estática, Cartas de revisión física (2023). DOI:10.1103/PhysRevLett.131.243003
Información de la revista: Cartas de revisión física
Proporcionado por la Universidad de Chicago