Investigadores de Skoltech y MIPT y sus colegas alemanes, austriacos y noruegos han propuesto y probado un nuevo método para modelar por ordenador aleaciones magnéticas. El método, que se basa en el aprendizaje automático, predijo con precisión las características energéticas, mecánicas y magnéticas de la aleación de hierro y aluminio.
Esto ha sido posible teniendo en cuenta los llamados momentos magnéticos de los átomos que dan lugar a los efectos del magnetismo. El estudio se publica en Scientific Reports y es un trampolín hacia el modelado de nitruro de cromo, un material ultraduro y resistente a la corrosión utilizado en la formación de metales, herramientas médicas e implantes.
El modelado informático de materiales suele ser un acto de equilibrio entre velocidad y precisión. El estándar de oro para predecir la estructura y las propiedades de los materiales con el menor error son los cálculos de la mecánica cuántica, como la resolución de la ecuación de Schrodinger.
Hay formas de acelerar estos exigentes cálculos, siendo la más popular la teoría del funcional de densidad. La forma en que la DFT ahorra tiempo de cálculo es la siguiente:en lugar de resolver la ecuación con respecto a la función de onda del electrón, encontramos la llamada densidad electrónica total en el estado de energía más bajo. Sin embargo, incluso eso sólo permite modelar en una supercomputadora sistemas de decenas o cientos de átomos de tamaño.
Los sistemas más grandes requieren una mayor simplificación:ignorar la estructura electrónica y considerar los llamados potenciales de interacción interatómica, que caracterizan las fuerzas entre los átomos. Naturalmente, esto sacrifica cierta precisión a la hora de predecir las propiedades de un material.
En los últimos años se ha visto el surgimiento de una nueva solución que ofrece lo mejor de ambos mundos. Conserva la precisión de los cálculos de la mecánica cuántica y aumenta drásticamente la velocidad de cálculo incluso para sistemas que suman miles de átomos. Un enfoque popular es utilizar el aprendizaje automático para obtener potenciales interatómicos entrenados en resultados de cálculos de mecánica cuántica.
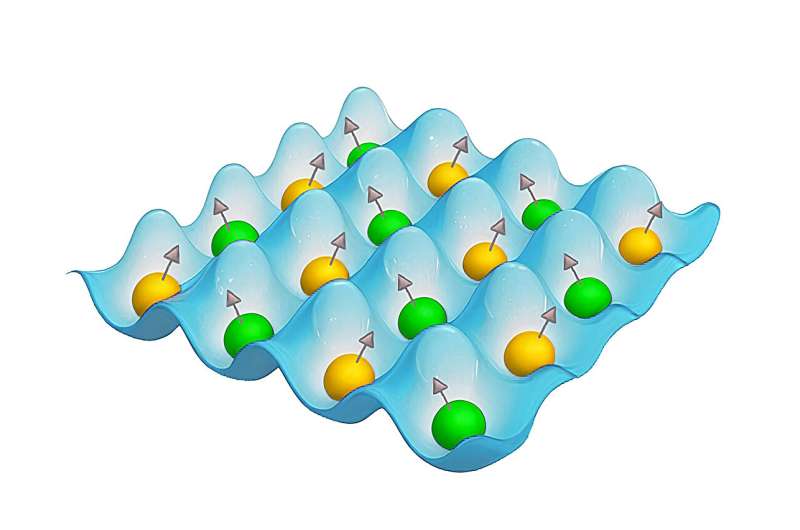
Estos potenciales ofrecen mejores predicciones de las propiedades de los materiales que sus análogos obtenidos experimentalmente. Sin embargo, los potenciales interatómicos del aprendizaje automático no necesariamente tienen en cuenta los momentos magnéticos de los átomos, y esto puede provocar errores en el modelado de materiales magnéticos.
Para modelar las propiedades de dichos materiales, un grupo de físicos y matemáticos de MIPT y Skoltech actualizaron su método Moment Tensor Potentials para obtener potenciales interatómicos de aprendizaje automático, generalizándolo a la versión mMTP. Este nuevo MTP "magnético" ya se ha utilizado para predecir la energía del hierro en sus estados paramagnético y ferromagnético. El nuevo estudio en Scientific Reports aplica el método a la aleación de dos componentes de hierro y aluminio.
Ivan Novikov, científico investigador senior de Skoltech y profesor asociado del Departamento de Física Química de Materiales Funcionales del MIPT, comentó:"Nuestro equipo está desarrollando potenciales de aprendizaje automático que aceleran los cálculos de la mecánica cuántica necesarios para describir las propiedades de los materiales en aproximadamente cinco órdenes de magnitud.
"Durante los últimos tres años, han ido surgiendo potenciales de aprendizaje automático con momento magnético, y creamos nuestro propio mMTP y lo validamos en el sistema de hierro. En el nuevo artículo, buscamos validar el potencial en un sistema de dos componentes y demostrar el algoritmo para construir un conjunto de datos para entrenar el potencial."
Los investigadores compilaron el conjunto de datos basándose en cálculos de mecánica cuántica y lo utilizaron para entrenar cinco mMTP. Luego, el equipo probó qué tan bien los potenciales podían predecir la estructura y las propiedades magnéticas de la aleación de hierro y aluminio dependiendo de la proporción de aluminio.
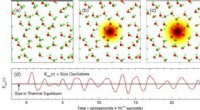
La primera etapa del estudio, que duró más tiempo, implicó la creación del conjunto de datos para el entrenamiento del modelo. Para los cálculos de mecánica cuántica se eligieron sistemas de dieciséis átomos. Los sistemas diferían en el número y las posiciones relativas de los átomos de hierro y aluminio. Para cada configuración, la teoría funcional de la densidad permitió al equipo encontrar las posiciones de los átomos, la geometría de la red y los momentos magnéticos que correspondían al estado de energía más bajo de ese sistema en particular.
A continuación, los investigadores introdujeron perturbaciones en el sistema desplazando posiciones atómicas y extendiendo o comprimiendo los vectores reticulares, que caracterizan la geometría reticular. La etapa final implicó excitar los momentos magnéticos de las estructuras tanto de la primera como de la segunda etapa utilizando la teoría funcional de la densidad y las restricciones que impone a los momentos magnéticos. El conjunto de datos resultante contenía más de 2000 configuraciones, tanto excitadas como en estado de equilibrio.
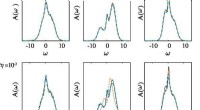
Luego, los científicos procedieron a entrenar un conjunto de cinco mMTP en el conjunto de datos recién formado y probaron sus predicciones de los momentos magnéticos de equilibrio de una configuración y los vectores reticulares frente a cálculos de la mecánica cuántica. El nuevo método demostró ser muy preciso independientemente de la proporción de aluminio en la aleación.
Las predicciones de MTP magnético también coincidieron bien con el experimento. Los investigadores consideraron cómo la proporción entre los metales de la aleación de hierro y aluminio afecta a los vectores reticulares. Resultó que la geometría de la red se mantuvo sin cambios para una proporción de aluminio entre 20% y 40%. Se observó una discrepancia cuantitativa, pero podría explicarse por el hecho de que el modelo asumió el cero absoluto de temperatura, a diferencia del experimento.
Los científicos compararon los momentos magnéticos de las aleaciones obtenidos mediante mMTP y mediante cálculos de mecánica cuántica. Los valores coincidían entre sí y con la teoría:a medida que aumentaba la proporción de aluminio, disminuían las propiedades magnéticas de la aleación. Pero mientras que el mMTP predijo una pérdida completa de ferromagnetismo con un 50% de aluminio, los cálculos de la mecánica cuántica no lo hicieron. Esta discrepancia exige una mayor investigación.
Los investigadores planean complementar su método con aprendizaje activo, de modo que la selección de las configuraciones adecuadas para entrenar el potencial se realice automáticamente. Esto permitirá estudiar sistemas y materiales paramagnéticos multicomponentes a temperaturas distintas de cero.
"Combinando nuestro conocimiento y los hallazgos de la investigación de 2022 sobre el hierro con este nuevo artículo sobre la aleación de hierro y aluminio, agregaremos aprendizaje activo y verificaremos mMTP en otro material:el nitruro de cromo", dijo Novikov.
"Específicamente, podremos predecir la variación de la capacidad calorífica específica y examinar los estados paramagnéticos. Estoy a favor del enfoque cuando se comienza por validar exhaustivamente el método y sólo después se pasa a cuestiones prácticas. Y este es el camino Nuestra investigación ha avanzado hasta ahora:primero validamos MTP en sistemas de referencia y ahora estamos en un punto en el que podemos comenzar a predecir los diagramas de fases de materiales más complejos".
Más información: Alexey S. Kotykhov et al, Potenciales de aprendizaje automático magnéticos basados en DFT restringidos para aleaciones magnéticas:un estudio de caso de Fe-Al, Scientific Reports (2023). DOI:10.1038/s41598-023-46951-x
Información de la revista: Informes científicos
Proporcionado por el Instituto de Ciencia y Tecnología de Skolkovo