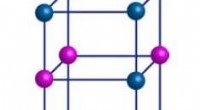
Modelo estructural de fibrilla de péptido amiloide A-beta 1-42 de Alzheimer derivada de una estructura experimental (PDB:2MXU). Los agregados fibrilares actúan como toxinas celulares al inicio y la progresión de la enfermedad de Alzheimer. Crédito:Emanuel Peter
Proteínas los omnipresentes caballos de batalla de la bioquímica, son moléculas enormes cuya función depende de cómo se pliegan en estructuras intrincadas. Para comprender cómo funcionan estas moléculas, los investigadores utilizan modelos informáticos para calcular cómo se pliegan las proteínas.
Ahora, un nuevo algoritmo puede acelerar esas simulaciones vitales, permitiéndoles modelar fenómenos que antes estaban fuera de su alcance. Los resultados pueden eventualmente ayudar a los científicos a comprender y tratar mejor enfermedades como el Alzheimer, dijo Emanuel Peter, químico de la Universidad de Ratisbona. Su trabajo sobre la nueva técnica se describe esta semana en La Revista de Física Química .
Simulaciones convencionales, utilizando dinámica molecular y métodos de Monte Carlo, han tenido éxito en general en el modelado de moléculas biológicas como proteínas. Para determinar cómo se pliegan las proteínas, la simulación busca configuraciones que correspondan a estados de menor y menor energía. Finalmente, encuentra el estado de energía más bajo, lo que le da un pliegue estable. Pero a medida que busca la simulación, puede encontrar una configuración con una energía ligeramente superior, que forma una barrera que impide el algoritmo.
Como resultado de estas ralentizaciones, Los métodos convencionales solo pueden simular comportamientos moleculares que ocurren en escalas de tiempo cortas de unos pocos cientos de microsegundos. Muchos fenómenos, como ciertos pliegues de proteínas o un fármaco que se une a un objetivo potencial, suceden en el transcurso de unos segundos, minutos o incluso días. Simular escalas de tiempo tan largas llevaría demasiado tiempo de cálculo con solo enfoques convencionales.
Para acelerar las simulaciones, los investigadores pueden inyectar energía en el sistema, lo que empuja al modelo por encima de cualquier barrera energética. Pero uno de los mayores desafíos para estos métodos es definir las coordenadas que describen el sistema, que, por ejemplo, puede ser la longitud entre átomos en la molécula, y los ángulos entre enlaces. Tradicionalmente, los investigadores definen las coordenadas antes de iniciar la simulación. Cada paso de tiempo a lo largo de cada coordenada depende del paso anterior. Pero esta dependencia puede sesgar la simulación.
El nuevo algoritmo de Peter evita este sesgo. Encontró un sistema de coordenadas generalizado en el que cada paso de tiempo no se basa en el paso anterior. "Solo se necesitan unos pocos parámetros, y no se requiere intuición humana, que potencialmente puede sesgar el resultado de la simulación, " él dijo.
Para probar el nuevo algoritmo, Peter lo usó para modelar el agua, un péptido llamado dialanina, el plegamiento de otro péptido llamado TrpCage, y la aglutinación de beta amiloide 25-35, que son fragmentos de proteínas asociados con la enfermedad de Alzheimer. En cada caso, su técnica informa haber acelerado las simulaciones. Y las simulaciones de beta amiloide podrían ayudar a explicar por qué la enfermedad de Alzheimer ha sido difícil de tratar.
En la enfermedad de Alzheimer, Los fragmentos de proteína beta-amiloide se agregan, formando una placa dura que se acumula entre las neuronas y las interrumpe. La beta amiloide también es una toxina, que conduce a la muerte de las células neuronales y la degeneración de la función neuronal. Las nuevas simulaciones sugieren que la beta amiloide puede asumir una variedad de estructuras. Esta flexibilidad estructural podría ser la razón por la que algunos medicamentos que intentan inhibir la agregación no hayan tenido éxito, Dijo Peter. Cuando esos medicamentos se unen a la beta amiloide, la beta amiloide simplemente cambia de forma, permitiendo que continúe aglutinando. El fármaco se incorpora al agregado y la placa.
Este tipo de flexibilidad estructural, llamada entropía de conformación, también es una característica clave en otros péptidos que forman placas tóxicas en enfermedades como la enfermedad de Huntington, Diabetes tipo 2, y enfermedad de Parkinson. Por lo tanto, el nuevo algoritmo también podría ser útil para comprender estas otras enfermedades.