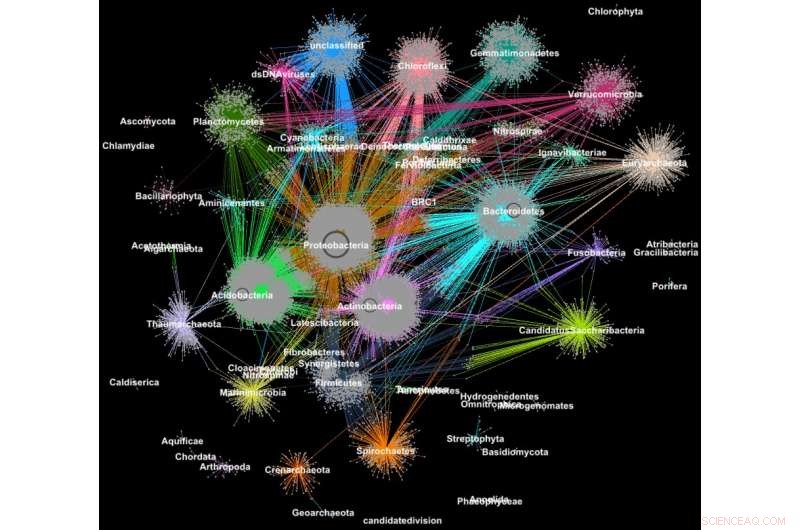
Las proteínas de los metagenomas se agruparon en familias según su clasificación taxonómica. Crédito:Georgios Pavlopoulos y Nikos Kyrpides, Laboratorio de JGI / Berkeley
¿Sabía que las herramientas que se utilizan para analizar las relaciones entre los usuarios de redes sociales o clasificar las páginas web también pueden ser extremadamente valiosas para dar sentido a los grandes datos científicos? En una red social como Facebook, cada usuario (persona u organización) se representa como un nodo y las conexiones (relaciones e interacciones) entre ellos se denominan bordes. Al analizar estas conexiones, los investigadores pueden aprender mucho sobre cada usuario:intereses, aficiones, hábitos de compra, amigos, etc.
En biología, Se pueden usar algoritmos similares de agrupamiento de gráficos para comprender las proteínas que realizan la mayoría de las funciones de la vida. Se estima que solo el cuerpo humano contiene alrededor de 100, 000 tipos de proteínas diferentes, y casi todas las tareas biológicas, desde la digestión hasta la inmunidad, ocurren cuando estos microorganismos interactúan entre sí. Una mejor comprensión de estas redes podría ayudar a los investigadores a determinar la efectividad de un medicamento o identificar tratamientos potenciales para una variedad de enfermedades.
Hoy dia, Las tecnologías avanzadas de alto rendimiento permiten a los investigadores capturar cientos de millones de proteínas, genes y otros componentes celulares a la vez y en una variedad de condiciones ambientales. Luego, se aplican algoritmos de agrupación en clústeres a estos conjuntos de datos para identificar patrones y relaciones que pueden apuntar a similitudes estructurales y funcionales. Aunque estas técnicas se han utilizado ampliamente durante más de una década, no pueden seguir el ritmo del torrente de datos biológicos que generan los secuenciadores y microarrays de próxima generación. De hecho, muy pocos algoritmos existentes pueden agrupar una red biológica que contenga millones de nodos (proteínas) y bordes (conexiones).
Es por eso que un equipo de investigadores del Laboratorio Nacional Lawrence Berkeley (Berkeley Lab) y del Joint Genome Institute (JGI) del Departamento de Energía (DOE) adoptó uno de los enfoques de agrupamiento más populares en la biología moderna:el algoritmo de agrupamiento de Markov (MCL), y lo modificó para que se ejecute rápidamente, de manera eficiente y a escala en supercomputadoras de memoria distribuida. En un caso de prueba, su algoritmo de alto rendimiento, llamado HipMCL, logró una hazaña previamente imposible:agrupar una gran red biológica que contiene alrededor de 70 millones de nodos y 68 mil millones de bordes en un par de horas, usando aproximadamente 140, 000 núcleos de procesador en la supercomputadora Cori del Centro Nacional de Investigación en Computación Científica (NERSC). Recientemente se publicó en la revista un artículo que describe este trabajo. Investigación de ácidos nucleicos .
"El beneficio real de HipMCL es su capacidad para agrupar redes biológicas masivas que eran imposibles de agrupar con el software MCL existente, lo que nos permite identificar y caracterizar el espacio funcional novedoso presente en las comunidades microbianas, "dice Nikos Kyrpides, quien dirige los esfuerzos de ciencia de datos de microbiomas de JGI y el programa Prokaryote Super y es coautor del artículo. "Además, podemos hacerlo sin sacrificar la sensibilidad o precisión del método original, que es siempre el mayor desafío en este tipo de esfuerzos de escala ".
"A medida que crecen nuestros datos, Cada vez es más imperativo trasladar nuestras herramientas a entornos informáticos de alto rendimiento. ", agrega." Si me preguntaras qué tan grande es el espacio de proteínas? La verdad es, realmente no lo sabemos porque hasta ahora no teníamos las herramientas computacionales para agrupar de manera efectiva todos nuestros datos genómicos y sondear la materia oscura funcional ".
Además de los avances en la tecnología de recopilación de datos, Los investigadores optan cada vez más por compartir sus datos en bases de datos comunitarias como el sistema Integrated Microbial Genomes &Microbiomes (IMG / M), que se desarrolló a través de una colaboración de décadas entre científicos de JGI y la División de Investigación Computacional (CRD) de Berkeley Lab. Pero al permitir a los usuarios realizar análisis comparativos y explorar las capacidades funcionales de las comunidades microbianas en función de su secuencia metagenómica, herramientas comunitarias como IMG / M también están contribuyendo a la explosión de datos en tecnología.
Cómo los paseos aleatorios conducen a cuellos de botella informáticos
Para controlar este torrente de datos, los investigadores se basan en el análisis de conglomerados, o agrupación. Esta es esencialmente la tarea de agrupar objetos para que los elementos del mismo grupo (clúster) sean más similares que los de otros clústeres. Durante más de una década, Los biólogos computacionales han favorecido el MCL para agrupar proteínas por similitudes e interacciones.
"Una de las razones por las que MCL ha sido popular entre los biólogos computacionales es que está relativamente libre de parámetros; los usuarios no tienen que establecer una tonelada de parámetros para obtener resultados precisos y es notablemente estable a pequeñas alteraciones en los datos. Esto es importante porque es posible que deba volver a definir una similitud entre puntos de datos o que tenga que corregir un pequeño error de medición en sus datos. En estos casos, no desea que sus modificaciones cambien el análisis de 10 grupos a 1, 000 racimos, "dice Aydin Buluç, un científico del CRD y uno de los coautores del artículo.
Pero, él añade, la comunidad de biología computacional se encuentra con un cuello de botella informático porque la herramienta se ejecuta principalmente en un solo nodo de computadora, es computacionalmente costoso de ejecutar y tiene una gran huella de memoria, todo lo cual limita la cantidad de datos que este algoritmo puede agrupar.
Uno de los pasos de este análisis más intensivos desde el punto de vista computacional y de memoria es un proceso llamado recorrido aleatorio. Esta técnica cuantifica la fuerza de una conexión entre nodos, que es útil para clasificar y predecir enlaces en una red. En el caso de una búsqueda en Internet, esto puede ayudarlo a encontrar una habitación de hotel barata en San Francisco para las vacaciones de primavera e incluso indicarle cuál es el mejor momento para reservarla. En biología, una herramienta de este tipo podría ayudarlo a identificar las proteínas que ayudan a su cuerpo a combatir el virus de la influenza.
Dado un gráfico o una red arbitrarios, es difícil conocer la forma más eficiente de visitar todos los nodos y enlaces. Un paseo aleatorio da una idea de la huella al explorar todo el gráfico de forma aleatoria; comienza en un nodo y se mueve arbitrariamente a lo largo de un borde hasta un nodo vecino. Este proceso continúa hasta que se alcanzan todos los nodos de la red de gráficos. Debido a que hay muchas formas diferentes de viajar entre los nodos de una red, este paso se repite varias veces. Algoritmos como MCL continuarán ejecutando este proceso de caminata aleatoria hasta que ya no haya una diferencia significativa entre las iteraciones.
En cualquier red dada, es posible que tenga un nodo que esté conectado a cientos de nodos y otro nodo con una sola conexión. Las caminatas aleatorias capturarán los nodos altamente conectados porque se detectará una ruta diferente cada vez que se ejecute el proceso. Con esta información, el algoritmo puede predecir con cierto nivel de certeza cómo un nodo de la red está conectado a otro. Entre cada carrera de caminata aleatoria, el algoritmo marca su predicción para cada nodo del gráfico en una columna de una matriz de Markov, algo así como un libro mayor, y los grupos finales se revelan al final. Suena bastante simple pero para redes de proteínas con millones de nodos y miles de millones de bordes, esto puede convertirse en un problema extremadamente computacional y que requiere mucha memoria. Con HipMCL, Los científicos informáticos del Berkeley Lab utilizaron herramientas matemáticas de vanguardia para superar estas limitaciones.
"Hemos mantenido notablemente intacta la columna vertebral de MCL, haciendo de HipMCL una implementación masivamente paralela del algoritmo MCL original, "dice Ariful Azad, científico informático en CRD y autor principal del artículo.
Aunque ha habido intentos anteriores de paralelizar el algoritmo MCL para que se ejecute en una sola GPU, la herramienta solo podía agrupar redes relativamente pequeñas debido a limitaciones de memoria en una GPU, Azad señala.
"Con HipMCL, básicamente, reelaboramos los algoritmos de MCL para que se ejecuten de manera eficiente, en paralelo en miles de procesadores, y configúrelo para aprovechar la memoria agregada disponible en todos los nodos de cómputo, ", agrega." La escalabilidad sin precedentes de HipMCL proviene de su uso de algoritmos de última generación para la manipulación de matrices dispersas ".
Según Buluç, realizar una caminata aleatoria simultáneamente desde muchos nodos del gráfico se calcula mejor utilizando la multiplicación de matrices de matriz dispersa, que es una de las operaciones más básicas en el estándar GraphBLAS recientemente lanzado. Buluç y Azad desarrollaron algunos de los algoritmos paralelos más escalables para la multiplicación de matrices de matriz dispersa de GraphBLAS y modificaron uno de sus algoritmos de última generación para HipMCL.
"El quid aquí fue encontrar el equilibrio correcto entre el paralelismo y el consumo de memoria. HipMCL extrae dinámicamente tanto paralelismo como sea posible dada la memoria disponible asignada, "dice Buluç.
HipMCL:agrupación en clústeres a escala
Además de las innovaciones matemáticas, Otra ventaja de HipMCL es su capacidad para ejecutarse sin problemas en cualquier sistema, incluidas las computadoras portátiles, estaciones de trabajo y grandes supercomputadoras. Los investigadores lograron esto desarrollando sus herramientas en C ++ y utilizando bibliotecas MPI y OpenMP estándar.
"Probamos ampliamente HipMCL en Intel Haswell, Procesadores Ivy Bridge y Knights Landing en NERSC, usando hasta 2, 000 nodos y medio millón de hilos en todos los procesadores, y en todas estas ejecuciones, HipMCL agrupa con éxito redes que comprenden de miles a miles de millones de bordes, ", dice Buluç." Vemos que no existe una barrera en la cantidad de procesadores que puede utilizar para ejecutar y descubrir que puede agrupar redes 1, 000 veces más rápido que el algoritmo MCL original ".
"HipMCL será realmente transformador para la biología computacional de big data, al igual que los sistemas IMG e IMG / M han sido para la genómica del microbioma, ", dice Kyrpides." Este logro es un testimonio de los beneficios de la colaboración interdisciplinaria en Berkeley Lab. Como biólogos, entendemos la ciencia, pero ha sido muy valioso poder colaborar con científicos informáticos que pueden ayudarnos a abordar nuestras limitaciones e impulsarnos hacia adelante ".
Su siguiente paso es continuar reelaborando HipMCL y otras herramientas de biología computacional para futuros sistemas de exaescala, que podrá calcular quintillones de cálculos por segundo. Esto será esencial a medida que los datos genómicos continúen creciendo a un ritmo alucinante, duplicándose cada cinco o seis meses. Esto se hará como parte del centro de codiseño Exagraph del DOE Exascale Computing Project.