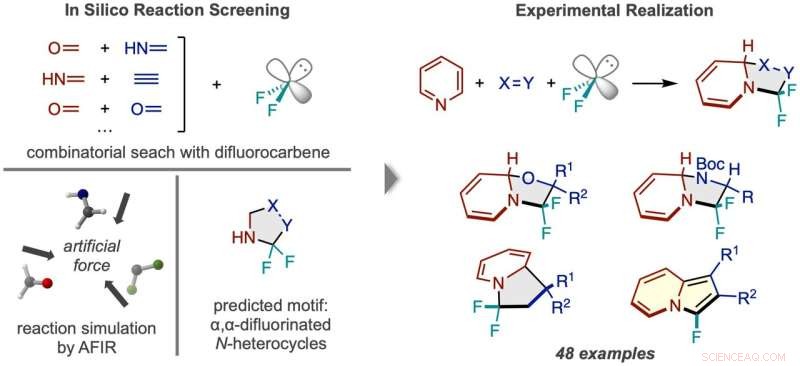
Flujo de trabajo del descubrimiento de reacciones mediante cribado in silico. (Izquierda) Se simularon reacciones entre el difluorocarbeno y numerosos pares de moléculas pequeñas, prediciendo un producto N-heterociclo fluorado dos veces en el carbono alfa. (Derecha) El marco de reacción exitoso usando piridina y ejemplos de los tipos de compuestos de productos obtenidos. Crédito:Síntesis de la naturaleza (2022). DOI:10.1038/s44160-022-00128-y
Las simulaciones por computadora se usan con mayor frecuencia como una guía para que los químicos puedan calcular de manera más eficiente los detalles exactos de una idea de reacción general que tienen en mente, al igual que una brújula ayuda a guiar a un explorador de manera eficiente a un destino en su mapa. Sin embargo, los investigadores de ICReDD llevaron las cosas un paso más allá y usaron simulaciones para producir la idea general de una reacción completamente inimaginable, usando efectivamente cálculos para hacer el mapa en sí. Utilizando el principio de diseño sugerido por los resultados computacionales, el equipo dio en el clavo en el laboratorio y desarrolló con éxito un conjunto de 48 reacciones que producen compuestos potencialmente útiles para el desarrollo de nuevos fármacos.
La presencia y posición del flúor en una molécula a menudo afecta la actividad farmacológica de una molécula. Los investigadores de ICReDD han utilizado cálculos químicos cuánticos para descubrir una reacción que agrega selectivamente dos átomos de flúor a una posición de difícil acceso en un N-heterociclo:moléculas con una estructura de anillo de carbono donde al menos un carbono en el anillo se reemplaza con nitrógeno. . La capacidad de unir átomos de flúor al "carbono alfa" de difícil acceso, el carbono inmediatamente al lado del nitrógeno en la estructura del anillo, podría conducir al desarrollo de una gran cantidad de fármacos novedosos.
Antes de llevar a cabo experimentos en el laboratorio, los investigadores lanzaron una amplia red, probando computacionalmente la viabilidad de numerosas reacciones de 3 componentes utilizando el método de reacción inducida por fuerza artificial (AFIR). Simularon la reacción de una molécula de difluorocarbeno, que actúa en la fuente de los átomos de flúor, con varios pares de pequeñas moléculas que presentan un doble o triple enlace. Estas simulaciones mostraron que una serie de reacciones de formación de anillos deberían ser viables.
Los investigadores probaron una de las reacciones prometedoras sugeridas por los resultados computacionales iniciales, pero no tuvieron éxito. A more narrowly focused, optimized computation of the transition state energy of the reaction in question showed that the difluorocarbene molecule more easily reacted with itself than with the desired starting molecules, signaling that an undesired side reaction was likely occurring. This result inspired researchers to change one of the starting materials to the cyclic molecule pyridine, which they anticipated would be able to compete with the unwanted side reaction. This change resulted in the successful synthesis of the desired N-heterocyclic product with two fluorines attached at the alpha carbon position.
The reaction developed here is also significant because it breaks the aromatic system of electrons in the pyridine molecule, a transformation that is especially difficult due to the high stability of aromatic systems. Additionally, the 3-component reaction framework was applied successfully in the lab to a wide range of starting materials, resulting in many new molecules with unique alpha position fluorine substitutions. The large scope of reactivity greatly increases the potential utility of this reaction framework in new drug development.
The researchers see their streamlined screening method as a way to broaden the scope of their search and discover new horizons in chemical reaction design.
"Our study's highlight is the successful demonstration of an in silico reaction screening strategy for reaction development. The computational reaction simulation suggested less-explored three-component reactions of difluorocarbene and two unsaturated molecules, which we successfully realized in experiments," explained lead author Hiroki Hayashi.
"I think the AFIR method is a powerful tool for dictating new research directions in reaction discovery, and we plan to continue building a computation-based reaction development platform by integrating the computational and informatics techniques of ICReDD."
The study was published in Nature Synthesis . Hitting rewind to predict multi-step chemical reactions