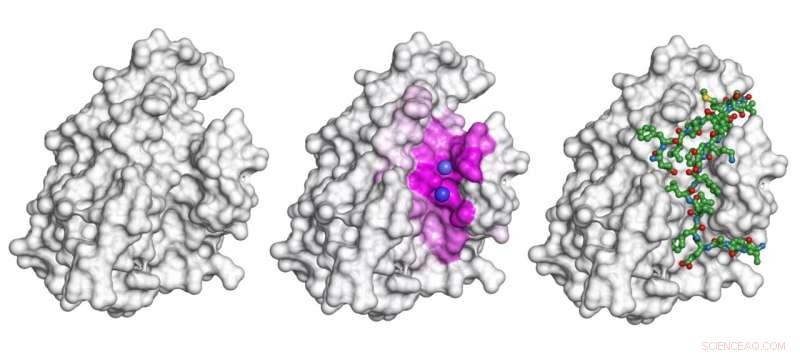
La forma gris es una proteína. Para el escenario de esta proteína que se une al péptido que se muestra como un modelo verdoso de palo y bola a la derecha, el modelo presentado en el estudio destaca la superficie involucrada en la interacción (el área rosa en el medio) y predice los sitios de unión exactos (esferas violetas). Crédito:Igor Kozlovskii y Petr Popov / Skoltech
Dos investigadores de Skoltech han presentado un modelo de red neuronal altamente eficiente que utiliza datos sobre la estructura de las proteínas para predecir cuáles de sus partes interactúan con otras moléculas biológicas llamadas péptidos. Saber esto es útil para desarrollar fármacos basados en péptidos, que puede afectar las interacciones proteína-proteína dentro de las células de una manera dirigida y no tóxica, regulando una amplia gama de procesos celulares. El estudio salió en el Revista de información y modelado químico .
Las proteínas son la maquinaria de las células, moviendose, comprometerse entre sí, y ejecutando todo tipo de operaciones. Los farmacólogos siempre han estado intrigados por la perspectiva de jugar con las interacciones entre proteínas. Sin embargo, parecían estar fuera de los límites como posible objetivo farmacológico:las moléculas terapéuticas más grandes, llamados biológicos, no podía penetrar en la célula para actuar sobre las proteínas, mientras que los agentes de molécula pequeña a menudo demostraron ser incapaces de tal acción.
Péptidos que median o regulan naturalmente alrededor del 40% de los procesos celulares, ocupan un terreno intermedio prometedor y tienen perspectivas de medicamentos dirigidos a las interacciones proteína-proteína. Los péptidos ofrecen lo mejor de ambos mundos:como moléculas pequeñas, pueden penetrar la membrana celular para alcanzar sus objetivos, y también exhiben baja toxicidad, junto con una alta afinidad y especificidad (acción fuerte y enfocada), las características de los productos biológicos.
Para diseñar fármacos basados en péptidos, Los farmacólogos necesitan conocer los llamados sitios de unión para cualquier proteína diana dada. Es decir, las manchas de la proteína que pueden unirse a un péptido. Cuantos más sitios de este tipo se conozcan, cuantas más oportunidades haya disponibles para el diseño de fármacos.
Los investigadores pueden identificar los sitios de unión de forma experimental, por ejemplo, utilizando cristalografía de rayos X, que revela la estructura 3D de las proteínas cristalizadas al estudiar cómo difractan los rayos X. Pero esto es muy caro para una larga lista de moléculas, y los métodos computacionales ofrecen una alternativa más rápida y económica. Algunos de ellos se basan en técnicas de aprendizaje automático, y a medida que se acumulan más datos sobre las estructuras de los complejos proteína-péptido, estos métodos se vuelven más poderosos y ofrecen predicciones de sitios de enlace cada vez mejores.
En su artículo del 22 de julio en el Revista de información y modelado químico , Skoltech Ph.D. El estudiante Igor Kozlovskii y el profesor asistente Petr Popov del grupo iMolecule presentaron un método computacional llamado BiteNetPp, que aprovecha el poder de las redes neuronales convolucionales 3D para detectar sitios de unión proteína-péptido. En BiteNetPp, una estructura de proteína conocida se alimenta a una red neuronal, que luego destaca los sitios sospechosos de unión de péptidos, y genera un conjunto de coordenadas 3D putativas, junto con las puntuaciones de probabilidad asociadas.
Petr Popov comenta sobre el enfoque de la detección de sitios de unión como reconocimiento de imágenes, introducido originalmente en el artículo anterior del equipo y trasladado al estudio informado en esta historia:"Al igual que las redes neuronales se pueden entrenar para reconocer, decir, peatones o ciclistas en fotos ordinarias en 2D, Consideramos que la detección de sitios vinculantes es detectar un tipo particular de objeto en una imagen. La diferencia es que usamos datos de estructura atómica 3D como nuestras entradas, por lo que el modelo opera en 'voxels, "un análogo tridimensional de píxeles".
El modelo recién presentado en realidad se basa en el del artículo anterior. "Esto se llama adaptación de dominio. BiteNetPp es el primer modelo que se ha ajustado en un conjunto de datos de proteína-péptido después de haber sido entrenado inicialmente con datos de proteína-molécula pequeña, "Popov explica." Puedes imaginar esto como entrenar un modelo para identificar lugares donde los ciclistas tienden a detenerse en la calle, pero comienza con datos sobre dónde tienden a detenerse los peatones, y solo luego extiende su dominio a los ciclistas. En lugar de empezar desde cero, vuelves a entrenar al modelo, anticipando que los 'sitios de unión' para los ciclistas pueden compartir algunas similitudes con los que atraen a los peatones:ya sabes, puestos de helados, semáforos, esa clase de cosas."
Los creadores del modelo han demostrado que BiteNetPp supera constantemente los métodos de vanguardia existentes al comparar sus predicciones para los sitios de unión proteína-péptido que se conocen a través de observaciones experimentales. En tono rimbombante, el nuevo modelo tarda menos de un segundo en analizar la estructura de una única proteína, haciéndolo adecuado para estudios a gran escala. Hay miles de interacciones proteína-proteína que pueden ser dirigidas por fármacos basados en péptidos, por lo que los métodos computacionales deben ser lo suficientemente rápidos para que su detección sea factible en un contexto farmacológico.