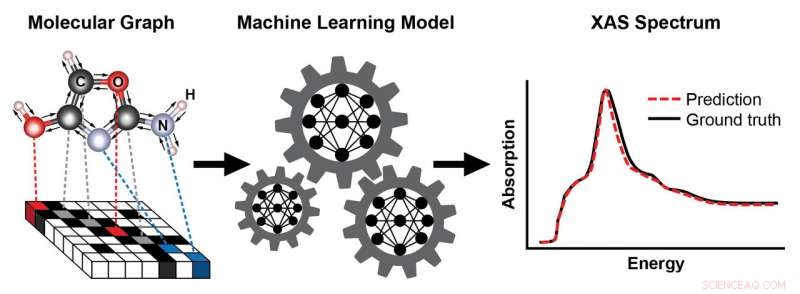
Un esquema que muestra los pasos para entrenar un modelo de aprendizaje automático para predecir un espectro de absorción de rayos X (XAS) basado en la estructura conocida de una molécula. La estructura de la molécula se representa como un gráfico, con átomos como nodos y enlaces químicos como aristas. Esta representación captura la conectividad de los átomos; aquí, carbono (C), oxígeno (O), nitrógeno (N), e hidrógeno (H), y el tipo y longitud de los enlaces químicos que los conectan. El espectro XAS resultante contiene una rica información sobre el entorno químico local de los átomos absorbentes, como su simetría y el número de átomos vecinos. Crédito:Laboratorio Nacional Brookhaven
La espectroscopia de absorción de rayos X (XAS) es una técnica de caracterización popular para sondear la estructura atómica local y las propiedades electrónicas de materiales y moléculas. Debido a que los átomos de cada elemento absorben rayos X a energías características, XAS es muy adecuado para trazar la distribución espacial de elementos en una muestra. Típicamente, Los científicos realizan experimentos XAS en fuentes de luz de sincrotrón, como la Fuente de luz de sincrotrón nacional II (NSLS-II), porque proporcionan una luz muy brillante, radiografías sintonizables. Midiendo la absorbancia en una muestra a diferentes energías de rayos X, los científicos pueden generar un gráfico llamado espectro de absorción de rayos X.
"XAS es una capacidad clave para los usuarios del NSLS-II del Laboratorio Nacional Brookhaven y el Centro de Nanomateriales Funcionales (CFN), Ambas instalaciones para usuarios de la Oficina de ciencia del Departamento de Energía de EE. UU. (DOE) que están abiertas a la comunidad de investigación científica, "dijo Deyu Lu, físico del Grupo de Teoría y Computación CFN. "Con las herramientas de análisis adecuadas, XAS puede proporcionar una gran comprensión de la investigación en nanociencia. El desarrollo de tales herramientas es fundamental para nuestra misión como instalaciones para el usuario ".
Clasificación de entornos químicos locales
Las diferentes regiones del espectro de absorción de rayos X son sensibles a diferentes aspectos de las propiedades del material en una muestra. Por ejemplo, la estructura de borde cercano de absorción de rayos X (XANES) se centra en la región del borde cercano del espectro, justo encima de la energía de inicio suficiente para excitar un electrón de las capas internas de un átomo a un estado vacío. XANES codifica información rica sobre el entorno químico local de los átomos absorbentes en una muestra, incluida su coordinación geométrica, simetría, y estado de carga (el número de electrones ganados o perdidos por enlaces químicos). Pero analizar los datos espectrales es un gran desafío debido a su naturaleza abstracta.
"A diferencia de una imagen de microscopio de un material donde se pueden ver directamente características como cristalinidad o defectos, Los espectros XANES codifican información que requiere experiencia en el dominio para interpretar, "explicó Lu.
La interpretación estándar de señales en un espectro XANES se basa en rasgos característicos conocidos como "huellas dactilares, "que se construyen a partir de mediciones en materiales de referencia. Sin embargo, este enfoque de huellas dactilares falla cuando la muestra no es un simple cristal y los materiales de referencia pertinentes no pueden identificarse fácilmente.
Las simulaciones teóricas a gran escala a partir de modelos de estructura atómica pueden proporcionar conocimientos muy útiles para la interpretación de espectros XANES experimentales; sin embargo, estas simulaciones suelen ser costosas desde el punto de vista computacional y requieren mucho tiempo, y su nivel de precisión depende en gran medida de las aproximaciones teóricas elegidas y del sistema en estudio. Como resultado, La interpretación espectral robusta es actualmente el cuello de botella de los estudios XAS. Es más, La interpretación en tiempo real de los espectros XAS ha surgido como un nuevo desafío para los estudios de la evolución dinámica de materiales en condiciones operativas y experimentación autónoma. La necesidad de robustez, La interpretación espectral eficiente se está generalizando cada vez más en las fuentes de luz de sincrotrón.
"Tiempo real, interpretación precisa de la dispersión de rayos X y las mediciones de espectroscopía, como la absorción de rayos X, fluorescencia, y la difracción es una capacidad importante para los usuarios que realizan investigaciones en NSLS-II y otras instalaciones de luz de sincrotrón, "dijo Mehmet Topsakal, un científico asociado en el Grupo de Materiales para Aplicaciones Energéticas del Departamento de Ciencia y Tecnología Nuclear de Brookhaven que está desarrollando técnicas avanzadas de análisis de datos y aprendizaje automático para espectroscopia de rayos X. "Todos los años, miles de científicos de todo el mundo vienen a NSLS-II para probar las propiedades de varios materiales. Una línea de análisis espectral de última generación permitiría a los usuarios obtener comentarios útiles sobre sus muestras mientras los experimentos están en curso y realizar ajustes sobre la marcha para guiar los experimentos. La pregunta es, ¿Cómo podemos hacer una interpretación espectral en tiempo real para descubrir correlaciones estructura-espectro? "
Extraer información con aprendizaje automático
Aprovechando el big data y el aprendizaje automático, Lu y Topsakal se propusieron responder a esta pregunta con el científico computacional Shinjae Yoo de la Iniciativa de Ciencia Computacional (CSI) de Brookhaven Lab y el Ph.D. de la Universidad de Columbia. Matthew Carbone, candidato y becario graduado en Ciencias Computacionales del DOE.
"La beca para graduados en ciencias computacionales del DOE me ha brindado una oportunidad única de extenderme más allá de mi investigación de doctorado en física química en Columbia para explorar el poder de los algoritmos de aprendizaje automático, trabajando junto a los científicos de Brookhaven, ", dijo Carbone." El aprendizaje automático aprovecha conjuntos de datos masivos para construir modelos altamente perceptivos que, una vez entrenado, puede hacer predicciones sobre la marcha sobre nuevos datos. Estos modelos podrían utilizarse para evitar los costosos cálculos de la química cuántica y respaldar la caracterización del material operativo ".
Los miembros de este equipo y los colaboradores han estado trabajando en mapeos de espectro a estructura y de estructura a espectro durante varios años. En 2017, desarrollaron modelos de aprendizaje automático para predecir los números de coordinación promedio de nanopartículas metálicas a partir de los espectros XANES. El año pasado, crearon una base de datos XANES para resolver la estructura local de un recubrimiento de óxido de titanio amorfo para aplicaciones fotocatalíticas. También construyeron un modelo de aprendizaje automático capaz de predecir la simetría local de los átomos absorbentes a partir de espectros XANES simulados de óxidos de metales de transición.
"Al realizar una interpretación espectral basada en la experiencia del dominio, tendemos a centrarnos en funciones específicas diseñadas a partir de nuestra intuición, ", dijo Lu." El aprendizaje automático puede extraer la información que necesitamos de una manera estadísticamente destacada que elimina el sesgo humano ".
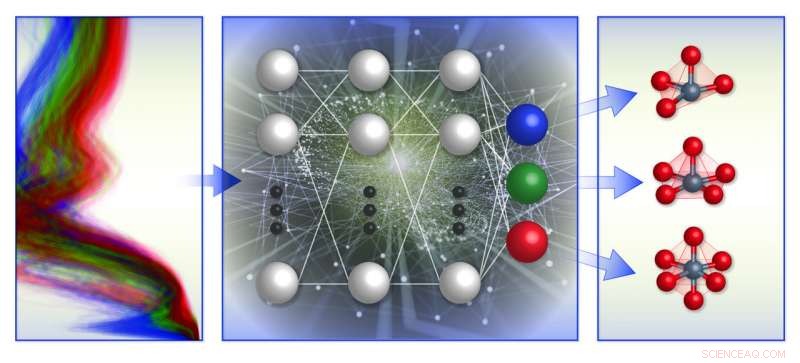
Una ilustración esquemática del marco de clasificación del entorno químico local basado en el espectro del equipo. Entrenaron modelos de aprendizaje automático (centro) con una base de datos de espectros de absorción de rayos X computacional (izquierda) para predecir la geometría local alrededor de iones de metales de transición cargados positivamente (derecha). Crédito:Laboratorio Nacional Brookhaven
Predicción de espectros de absorción de rayos X
Sobre la base de sus éxitos pasados, el equipo se enfrentó a un problema más desafiante:entrenar un modelo de aprendizaje automático para predecir rápidamente espectros basados en estructuras moleculares conocidas. Tal modelo evitaría la necesidad de simulaciones computacionalmente costosas, que no son factibles durante los experimentos de operando, cuando los científicos están estudiando materiales en condiciones operativas. A pesar de los crecientes esfuerzos de aprendizaje automático para predecir las propiedades químicas de los materiales, aún no se habían logrado predicciones directas de las funciones espectrales de materiales reales.
"Una dificultad técnica es construir una representación óptima de estructuras moleculares que pueda codificar la simetría inherente de las moléculas como características de entrada para el modelo de aprendizaje automático, "dijo Yoo.
Adoptando una idea reciente propuesta por científicos de Google, Topsakal y Carbone construyeron un modelo de aprendizaje automático basado en una representación gráfica de moléculas como entrada, donde los átomos se representan como nodos y los enlaces químicos como aristas.
"Las computadoras no pueden ver las moléculas como nosotros, "dijo Topsakal." Un gráfico es una forma natural de codificar la estructura y conectividad de una molécula, capturando qué átomos están conectados y el tipo y longitud de los enlaces químicos que los conectan. Es más, esta representación es invariante a transformaciones como traslaciones y rotaciones. Este concepto es análogo al del reconocimiento de imágenes, donde un objeto como un gato o un perro en un fondo aún se puede clasificar correctamente después de que la imagen se haya transformado ".
Para entrenar el modelo para una demostración de prueba de principio, el equipo utilizó una base de datos bien establecida (llamada QM9) que contiene información estructural y química calculada sobre 134, 000 moléculas pequeñas con hasta nueve átomos pesados por tipo de átomo (carbono, nitrógeno, oxígeno, y flúor). De esta base de datos, seleccionaron dos subconjuntos de entrenamiento:un subconjunto con moléculas que contienen al menos un átomo de oxígeno, y otro subconjunto con moléculas que contienen al menos un átomo de nitrógeno, y calcularon sus correspondientes espectros XANES. Luego, utilizaron sus modelos entrenados para predecir los espectros XANES de los bordes de absorción de oxígeno y nitrógeno correspondientes a las excitaciones de los electrones en la capa más interna de los átomos respectivos.
El modelo de aprendizaje automático reprodujo casi todos los picos de absorción significativos y predijo las posiciones de los picos (energías en las que aparecen los picos) y las alturas (intensidades de absorción) con una precisión muy alta. El modelo también recogió automáticamente el conocimiento del dominio de que la espectroscopia de absorción de rayos X es sensible a los grupos funcionales, o grupos de átomos con propiedades químicas y reactividad similares. Dependiendo del grupo funcional al que pertenezca el átomo absorbente, aparecen diferentes características en los espectros.
"Somos los primeros en demostrar que un modelo de aprendizaje automático se puede utilizar para predecir con precisión funciones espectrales completas de sistemas físicos reales directamente desde sus estructuras, ", dijo Topsakal." Aunque nos centramos en la espectroscopia de absorción de rayos X en nuestro estudio, este método podría generalizarse para predecir información espectral para otras técnicas populares, incluida la espectroscopia de rayos gamma e infrarrojos ".
"Una vez que entrenamos el modelo de aprendizaje automático, no necesitamos ejecutar simulaciones físicas que consumen mucho tiempo, que toman minutos, horas, o incluso días, ", dijo Yoo." No solo permitimos la predicción de espectros en tiempo real, sino también la generación simultánea de cientos y miles de inferencias de espectros mediante el uso de múltiples unidades de procesamiento de gráficos, o GPU. Dicha tecnología es clave para permitir controles automatizados de líneas de luz y acelerar el descubrimiento científico. Combinado con métodos para muestrear estructuras de materiales, estos modelos se pueden utilizar para seleccionar rápidamente estructuras relevantes para impulsar el diseño y el descubrimiento de materiales ".
Próximo, al equipo le gustaría combinar conceptos de su modelo que predice la simetría local a partir de los espectros XANES y este nuevo modelo que predice los espectros XANES a partir de estructuras moleculares. Por último, su objetivo es extraer información más completa sobre el entorno químico local o incluso la estructura de moléculas completas a partir de mediciones experimentales.
"Herramientas de aprendizaje automático, como los de reconocimiento de imágenes y voz y descubrimiento de fármacos, están en rápido desarrollo, ", dijo Lu." La clave es descubrir cómo adaptar estas herramientas de una manera innovadora para abordar los problemas de la ciencia de los materiales ".
"Nuestro objetivo en el desarrollo de tecnologías de inteligencia artificial y aprendizaje automático es resolver desafíos científicos únicos mediante la adopción de los últimos avances tecnológicos en estas áreas y la creación de enfoques novedosos que contribuyan a las respectivas comunidades de investigación, "añadió Yoo.