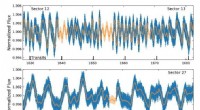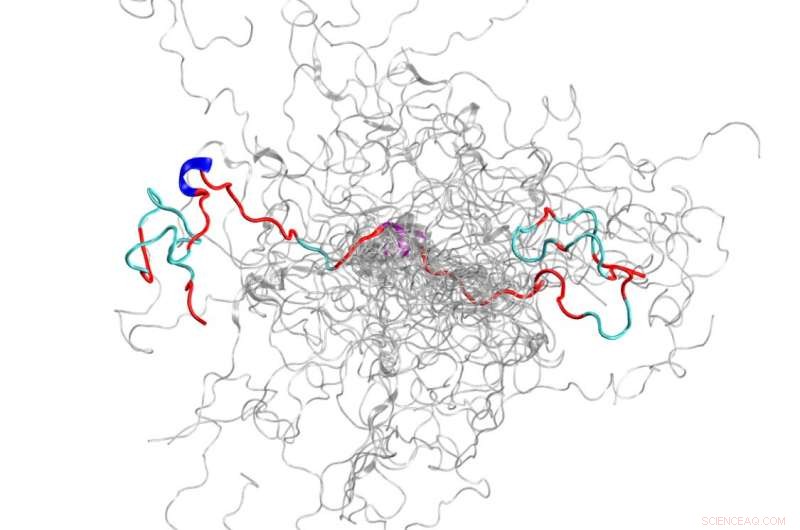
El conjunto configuracional (una colección de estructuras 3D) de una proteína intrínsecamente desordenada, el N-terminal de c-Src quinasa, que es una proteína de señalización importante en los seres humanos. Crédito:Laboratorio Nacional Oak Ridge, Departamento de Energía de EE. UU.
Usando la supercomputadora Titan y la fuente de neutrones de espalación en el Laboratorio Nacional de Oak Ridge del Departamento de Energía, Los científicos han creado el modelo tridimensional más preciso hasta ahora de una proteína intrínsecamente desordenada, revelando el conjunto de sus estructuras a nivel atómico.
Como su nombre lo indica, un IDP no adopta una orden, estructura estática como otras proteínas; en lugar de, es flexible y puede adoptar múltiples estructuras 3D. Esta falta de una estructura única es necesaria para la función biológica de los desplazados internos, pero dificulta su estudio desde el punto de vista técnico. Los desplazados internos pueden ser una proteína completa o un dominio de una proteína estructurada de otro modo, y constituyen una gran parte de humanos, microbio, y proteínas vegetales.
Loukas Petridis, un científico del personal en el Centro de Biofísica Molecular en ORNL, ha dirigido a un equipo de investigadores hacia una nueva forma de crear modelos físicos precisos de biosistemas tan flexibles, lo que puede conducir a una mejor comprensión de sus funciones biológicas. Durante los últimos tres años, el equipo ha combinado experimentos de dispersión de neutrones con simulaciones mejoradas de dinámica molecular de muestreo (MD) tan exigentes desde el punto de vista informático que requerían la potencia de procesamiento de Titán, el Cray XK7 de 27 petaflop recientemente retirado en las instalaciones de computación de liderazgo de Oak Ridge, una instalación para usuarios de la Oficina de Ciencias del DOE en ORNL.
"Estudiar a estos desplazados internos es bastante difícil, desde ambas perspectivas de experimentos y modelado, "dijo Utsab Shrestha, el autor principal del artículo del equipo, publicado recientemente en el procedimientos de la Academia Nacional de Ciencias . "No solo pensamos en ello a partir de experimentos o simulaciones, planeamos de manera que sinergizaríamos ambos enfoques, combinándolos de manera que pudiéramos obtener información más precisa sobre los desplazados internos. Específicamente, Las simulaciones nos ayudaron a generar un conjunto preciso de IDP a resolución atómica, lo cual es difícil de determinar únicamente a partir de experimentos ".
Típicamente, Los investigadores llevan a cabo experimentos como la dispersión de neutrones de ángulo pequeño, dispersión de rayos X de ángulo pequeño, o resonancia magnética nuclear para sondear sistemas biológicos flexibles. Sin embargo, Estos métodos no proporcionan una imagen detallada a nivel atómico de las estructuras tridimensionales de un desplazado interno, conocido como su conjunto configuracional. Es más, solo pueden producir datos promediados por conjuntos, en lugar de las configuraciones específicas de la estructura de la proteína subyacente. Los científicos también han realizado simulaciones por computadora de IDP y las han comparado con tales experimentos, con la esperanza de obtener los mismos resultados para verificar la precisión de sus modelos.
"Pero terminan por no estar de acuerdo con los experimentos, ", Dijo Petridis." Y debido a la discrepancia entre las simulaciones y los experimentos, tienen que volver a ponderar las simulaciones; tienen que ajustar los resultados de la simulación para que coincidan con los experimentos, lo cual es frustrante. Ese era el estado del arte hasta nuestro trabajo ".
Las simulaciones de MD por computadora realizadas por Shrestha utilizaron métodos de muestreo mejorados que lograron igualar no solo los experimentos de dispersión de neutrones, realizados por Viswanathan Gurumoorthy y sus colegas en SNS, una instalación para usuarios de la Oficina de Ciencias del DOE en ORNL, pero también datos NMR publicados anteriormente. Estas simulaciones de MD utilizan la física para determinar cómo se mueven las proteínas. La clave del éxito del equipo fue ejecutar muchas simulaciones de MD en paralelo en Titán, permitiendo que las simulaciones se comuniquen entre sí e intercambien información.
"Esto es muy importante porque permite que la simulación muestree un espacio configuracional más grande, explorar más de las estructuras tridimensionales de una manera más eficiente, "Dijo Petridis." Es por eso que este MD de muestreo mejorado puede producir resultados que la simulación de MD normal no puede producir. Tendríamos que ejecutar una simulación MD normal durante años para obtener los mismos resultados ".
El IDP que el equipo decidió estudiar es el dominio N-terminal de la c-Src quinasa, que es una proteína de señalización importante en los seres humanos. Las mutaciones en esta proteína compleja se han correlacionado con el cáncer, lo que también lo convierte en un importante objetivo farmacológico. Mientras mapea este dominio previamente turbio, los científicos pudieron descubrir nueva información sobre sus estructuras tridimensionales que los métodos anteriores no habían mostrado. Por ejemplo, aunque está en gran parte desordenado, esta proteína forma estructuras ordenadas transitorias, como hélices.
"La combinación de experimentos de dispersión de neutrones y simulación es muy poderosa, ", Dijo Petridis." La validación de las simulaciones en comparación con los experimentos de dispersión de neutrones es esencial para tener confianza en los resultados de la simulación. Las simulaciones validadas pueden proporcionar información detallada que no se obtiene directamente mediante experimentos ".
El modelo informático detallado del conjunto de estructuras tridimensionales del IDP abre la puerta a más experimentación. Por ejemplo, los científicos podrían simular el efecto de la fosforilación (la adición de un grupo fosfato a la proteína que puede regular la función de la proteína) para ver qué cambios estructurales tienen lugar en la c-Src quinasa que podrían influir en su función. También se podría examinar el papel de las mutaciones:si un investigador cambia un aminoácido en la cadena, ¿Cómo afecta esto a la estructura o al conjunto de estructuras?
"Hay muchas preguntas sin respuesta para la c-Src quinasa en particular que podrían responderse en términos de las interacciones con otros socios:el efecto de la fosforilación, el efecto de las mutaciones, "Dijo Petridis.
Más allá de los usos científicos potenciales del modelo en sí, Petridis ve oportunidades para aplicar el uso de la computación de alto rendimiento para ejecutar MD de muestreo mejorado para estudiar las estructuras de muchos otros desplazados internos importantes, que podría dar una idea de su función. Y más ampliamente, el equipo quiere desarrollar tecnologías de simulación que puedan reproducir perfiles de dispersión de neutrones de ángulo pequeño de sistemas biológicos aún más complejos.
"No queremos investigar solo las proteínas desordenadas, queremos tener sistemas mucho más grandes que contengan dominios ordenados y desordenados que puedan interactuar con membranas o ADN". "Dijo Petridis." La dispersión de neutrones es, en mi vista, la mejor técnica experimental para probar estos sistemas multicomponente, por ejemplo, una proteína que interactúa con una membrana o una proteína que interactúa con el ADN. Pero, todavía, la dispersión de neutrones necesita simulaciones precisas para interpretar mejor los datos ".