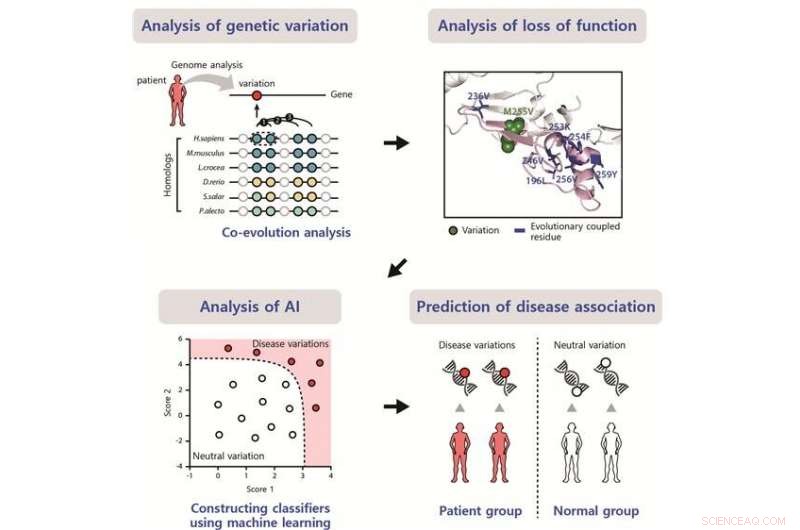
Esquema del método desarrollado para identificar el impacto de las variantes asociadas a la enfermedad. Crédito:POSTECH
Es importante predecir el impacto de las variantes de la secuencia de ADN para clasificar las variantes asociadas a la enfermedad (DV) de las variantes neutrales. Investigadores coreanos de la Universidad de ciencia y tecnología de Pohang (POSTECH) informan sobre el desarrollo de un método para predecir el impacto de la VD. El estudio aparece en la revista Investigación de ácidos nucleicos en junio.
Los métodos actuales para predecir los impactos mutacionales dependen de la conservación evolutiva en el sitio de la mutación, que se determina utilizando secuencias homólogas y se basa en la suposición de que las variantes en sitios bien conservados tienen un gran impacto. Sin embargo, muchos VD en sitios menos conservados pero funcionalmente importantes no pueden predecirse con los métodos actuales.
Los investigadores presentan un método para encontrar DV en sitios menos conservados mediante la predicción de los impactos mutacionales mediante el análisis de acoplamiento evolutivo. Los sitios funcionalmente importantes y evolutivamente acoplados a menudo tienen variantes compensatorias en sitios cooperativos para evitar la pérdida de función. Identificaron VD en sitios menos conservados que no se identificaron utilizando los métodos actuales basados en la conservación.
El profesor Kim dijo:"Este estudio se puede aplicar a una variedad de enfoques de la medicina de precisión, como el pronóstico de las enfermedades del paciente y la búsqueda de una medicina personalizada. Basado en un análisis de secuencia a gran escala, el método desarrollado es útil para encontrar más variantes asociadas a enfermedades que ayuden a encontrar biomarcadores y dianas terapéuticas de diversas enfermedades humanas ".