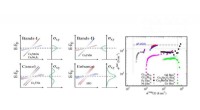
Las secuencias base del ácido nucleico, específicamente las que se encuentran en el ADN y el ARN, son herramientas invaluables en la clasificación filogenética por varias razones:
1. Lenguaje universal: Todos los organismos vivos utilizan el mismo código genético, lo que significa que las secuencias de ADN y ARN pueden compararse directamente en diferentes especies. Esto proporciona un lenguaje universal para comprender las relaciones evolutivas.
2. Mutación como cronometrador: Las mutaciones ocurren a una velocidad relativamente constante con el tiempo, actuando como un "reloj molecular". Al comparar el número de diferencias en las secuencias base entre dos especies, podemos estimar el tiempo ya que divergieron de un antepasado común.
3. Homología y similitud: Es probable que las secuencias que son similares o homólogas tengan ascendencia compartida. La comparación de secuencias nos permite identificar regiones que se han mantenido conservadas debido a su importancia funcional, proporcionando evidencia de relaciones evolutivas.
4. Diversos puntos de datos: Se pueden usar diferentes tipos de secuencias para el análisis filogenético, que incluyen:
* ADN nuclear: Ofrece información sobre la historia evolutiva general de los organismos.
* ADN mitocondrial (mtDNA): Evoluciona relativamente rápidamente, lo que lo hace útil para estudiar eventos evolutivos recientes y relaciones entre especies estrechamente relacionadas.
* ARN ribosómico (rRNA): Altamente conservado en diversos organismos, lo que lo hace valioso para estudiar relaciones evolutivas profundas.
5. Potencia computacional: Con el avance de las tecnologías de secuenciación y la bioinformática, podemos analizar grandes cantidades de datos de secuencia, generando árboles filogenéticos robustos basados en el análisis estadístico.
Cómo funciona en la práctica:
1. Colección de secuencias: Las secuencias de ADN o ARN se obtienen de las especies bajo investigación.
2. Alineación: Las secuencias están alineadas para identificar regiones homólogas y minimizar las diferencias debido a inserciones o deleciones.
3. Cálculo de distancia: El número de diferencias en las secuencias base entre especies se calcula para estimar la distancia evolutiva.
4. Construcción de árboles: Varios algoritmos (por ejemplo, unión de vecinos, máxima probabilidad) usan la información de distancia para construir árboles filogenéticos.
5. Interpretación: El árbol resultante representa las relaciones evolutivas entre las especies basadas en las similitudes y diferencias en sus secuencias genéticas.
Limitaciones:
* Variación de tasa: Las tasas de mutación pueden variar entre diferentes genes y especies, lo que lleva a inexactitudes en las estimaciones de tiempo.
* Transferencia de genes horizontales: La transferencia de material genético entre organismos no relacionados puede complicar la reconstrucción filogenética.
* Clasificación de linaje incompleto: La retención de polimorfismos ancestrales puede conducir a inconsistencias en la señal filogenética.
Conclusión:
Las secuencias de base de ácido nucleico juegan un papel crucial en la clasificación filogenética moderna, ofreciendo una herramienta poderosa para reconstruir las relaciones evolutivas y comprender la historia de la vida en la Tierra. Si bien existen limitaciones, el uso de estas secuencias ha revolucionado nuestra comprensión de la biodiversidad y la intrincada red de la vida.