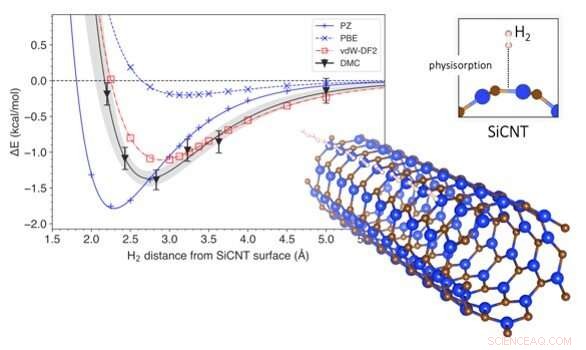
El cambio de energía asociado con la eliminación de hidrógeno de los nanotubos de carburo de silicio. El gráfico muestra la variación de la energía del sistema con la distancia de una molécula de hidrógeno a la superficie de un nanotubo de carburo de silicio (abajo a la derecha). La profundidad de la curva significa la energía necesaria para extraer hidrógeno del almacenamiento. Se presenta una comparación de métodos de predicción, siendo DMC el más preciso y vdW-DF2 su coincidencia más cercana. Crédito:Kenta Hongo de JAIST
La energía del hidrógeno tiene el potencial de ser una medida clave para alcanzar el objetivo de cero emisiones netas de las Naciones Unidas. pero su uso industrial se ha visto obstaculizado por la dificultad de su almacenamiento y manipulación. El hidrógeno se convierte en gas a muy baja temperatura (-252 ° C), lo que dificulta su almacenamiento a temperatura ambiente. La interacción entre el hidrógeno y su material de almacenamiento es simplemente demasiado débil para persistir a temperatura ambiente. Esto hace que el diseño de los materiales de almacenamiento sea crucial para lograr el objetivo de llevar la energía del hidrógeno al uso diario.
Aquí es donde entra en juego el diseño de materiales computacionales. Se puede ahorrar mucho tiempo y esfuerzo durante el desarrollo de la tecnología del hidrógeno diseñando un material en una computadora y simulando su capacidad de almacenamiento de hidrógeno. Pero las predicciones se vuelven muy limitadas en su uso a menos que sean precisas y puedan realizarse a un costo computacional razonable. En un estudio reciente publicado en ACS Omega , los científicos desarrollan un sistema computacionalmente costoso, pero un método novedoso y altamente preciso para predecir el almacenamiento de hidrógeno:"Mejorar la confiabilidad de la predicción para las simulaciones puede ayudar a acelerar el desarrollo de materiales para el almacenamiento de combustible de hidrógeno y conducir a una sociedad más eficiente desde el punto de vista energético, "dice el doctor Kenta Hongo del Instituto Avanzado de Ciencia y Tecnología de Japón (JAIST), quien dirigió el estudio.
Una de las fuerzas fundamentales de atracción entre objetos es la fuerza de van der Waals, que define la interacción entre átomos o moléculas en función de la distancia entre ellos. Dado que la fuerza de Van der Waals es la consecuencia de procesos cuánticos bastante complicados, los tratamientos convencionales no podrían describirlo bien, y por lo tanto, las simulaciones hasta ahora están al nivel de estimaciones aproximadas de la misma. Pero, ¿es correcto hacer eso al simular el almacenamiento de hidrógeno? Esta fue la principal preocupación del Dr. Hongo y su equipo.
Para responder a esta pregunta, miraron nanotubos de carburo de silicio, uno de los materiales más prometedores para el almacenamiento de hidrógeno. Usando una técnica computacional llamada difusión Monte Carlo (DMC), crearon un modelo que tenía en cuenta las fuerzas de van der Waals al simular el almacenamiento de hidrógeno en nanotubos de carburo de silicio. La mayoría de los modelos convencionales consideran las interacciones entre el hidrógeno y los nanotubos de carburo de silicio en su conjunto, pero el método DMC utiliza el poder de una supercomputadora para reconstruir fielmente el mecanismo de interacción siguiendo la disposición de los electrones individuales. Esto hace que el modelo DMC sea el método de predicción más preciso hasta la fecha. Usando el modelo DMC, los investigadores también pudieron predecir cuánta energía se requeriría para desalojar el hidrógeno de su almacenamiento, y qué tan lejos probablemente estaría el hidrógeno de la superficie del nanotubo de carburo de silicio. Luego compararon los resultados de su modelado con los obtenidos mediante métodos de predicción convencionales.
Los métodos de predicción convencionales se basan generalmente en técnicas computacionales llamadas teoría funcional de la densidad (DFT). DFT utiliza funcionales (descripciones de modelos de interacciones cuánticas) que describen las variaciones espaciales de la densidad de electrones para determinar las propiedades de sistemas complejos. Si bien se han realizado varios estudios basados en DFT sobre el almacenamiento de hidrógeno en nanotubos de carburo de silicio, ninguno de ellos ha incorporado las fuerzas de van der Waals en sus predicciones. Los funcionales DFT corregidos por Van der Waals tienen, sin embargo, ha sido empleado en la predicción de otros materiales. El Dr. Hongo y su equipo simularon el almacenamiento de hidrógeno utilizando una amplia gama de funciones DFT, los que tienen correcciones de van der Waals y los que no. Descubrieron que las funciones de DFT sin las correcciones de van der Waals desestimaron la energía requerida para el almacenamiento de hidrógeno en un 4–14%. Por otra parte, Las funciones de DFT corregidas por van der Waals produjeron resultados bastante similares a los de DMC. Es más, encontraron que la contribución de la fuerza de van der Waals a la energía de almacenamiento era aproximadamente del 9 al 29%, que es apenas insignificante.
Estos hallazgos, El Dr. Hongo cree, puede ser un trampolín para una mayor innovación en la tecnología de simulación de almacenamiento de hidrógeno. "Aunque el método DMC es computacionalmente costoso, se puede utilizar para aclarar las peculiaridades (tendencias de error de predicción) de cada método de predicción. Esto nos ayudará a comprender en qué predicción confiar, y también cómo modificar los métodos de predicción para hacerlos más útiles, " el explica.