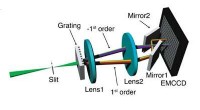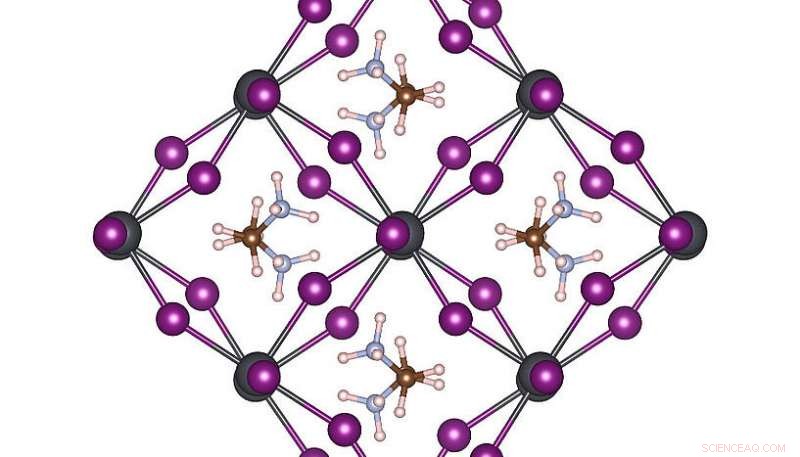
Estructura atómica de alta simetría de MAPbI3 a temperatura ambiente. Crédito:Menno Bokdam / Universidad de Viena
A escala atómica, los materiales pueden mostrar una rica paleta de comportamiento dinámico, que afecta directamente a las propiedades físicas de estos materiales. Durante muchos años, Ha sido un sueño describir estas dinámicas en materiales complejos a varias temperaturas usando simulaciones por computadora. Los físicos de la Universidad de Viena han desarrollado un método de aprendizaje automático sobre la marcha que permite dichos cálculos a través de la integración directa en el paquete de simulación Ab-initio de Viena (VASP) basado en la mecánica cuántica. La versatilidad del método de autoaprendizaje se demuestra con nuevos hallazgos, publicado en la revista Cartas de revisión física , en las transiciones de fase de las perovskitas híbridas. Estas perovskitas son de gran interés científico debido a su potencial en la captación de energía solar y otras aplicaciones.
A temperatura ambiente, todos los materiales se mueven constantemente a escala atómica. Incluso la roca sólida está formada por átomos que giran. Las propiedades físicas de los materiales están directamente relacionadas con la disposición de los átomos en el, así llamado red cristalina. Dependiendo de la temperatura o presión, esta disposición puede cambiar afectando las propiedades de los materiales. Uno puede pensar en el diamante que es transparente y duro debido a la disposición periódica de los átomos de carbono en el cristal de diamante. Los mismos átomos, arreglado de manera diferente, resultados en negro, grafito quebradizo. Ya era posible calcular con precisión las coordenadas de los átomos en materiales simples a diferentes temperaturas con simulaciones de dinámica molecular mecánica cuántica (MD). Sin embargo, Tales cálculos son computacionalmente costosos y restringen las aplicaciones prácticas a un par de cientos de átomos y un tiempo de simulación limitado.
Los físicos del grupo de Física de Materiales Computacionales de la Universidad de Viena han desarrollado un nuevo enfoque que supera estas limitaciones y hace posible la simulación de materiales complejos para futuras aplicaciones energéticas. Esto se logra desarrollando un algoritmo de autoaprendizaje basado en datos eficiente y robusto y, Más importante, integrando este algoritmo directamente en el paquete de simulación de Viena Ab-initio (VASP). En el nuevo enfoque, la "máquina" puede recoger, por sí mismo, los ingredientes esenciales para una descripción de modelo más simple de los átomos que interactúan durante las simulaciones de MD. Ya después de calcular algunos cientos de pasos de tiempo, la máquina puede predecir con suficiente precisión las posiciones de los átomos en el paso de tiempo consecutivo. La máquina también puede hacer una estimación de su precisión para los pasos consecutivos. Si el error es demasiado alto, la máquina cambia de marcha y realiza la muy precisa, pero caro, Cálculos de MD. Cuanto más tiempo de simulación pasa, cuanto más aprende la máquina y más precisa se vuelve. De este modo, se requieren cada vez menos cálculos de MD, lo que eventualmente conduce a la situación en la que la máquina realiza todos los pasos de tiempo. Es más, la capacidad de autoaprendizaje sobre la marcha reduce la necesidad de intervención humana requerida por otros métodos de aprendizaje automático existentes.
Para demostrar el poder de este nuevo método, los investigadores lo han aplicado para estudiar las transiciones entre diferentes estructuras atómicas del MAPbI 3 perovskita al cambiar la temperatura. Este material es muy popular debido a su potencial como un nuevo componente de celda solar económico. Está hecho de moléculas orgánicas que pueden girar rápidamente, separados entre sí por una red compuesta de átomos de plomo y yoduro. Dependiendo de la temperatura se forman tres fases cristalinas diferentes. Los mecanismos atómicos cercanos a la temperatura de transición son muy difíciles de determinar mediante experimentos, y las simulaciones MD requerirían años de tiempo de cálculo incluso en un sistema de supercomputación moderno. Después de aprender, la máquina puede predecir las temperaturas de transición de fase y las constantes de celosía de este material con una precisión sin precedentes. El método desarrollado es general y aplicable a muchos otros problemas futuros de ciencia de materiales y estará disponible para investigadores de todo el mundo en la próxima versión de VASP.
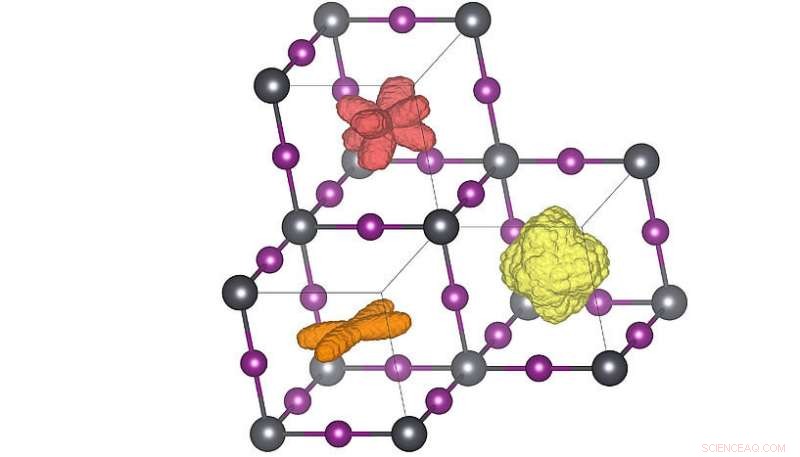
Distribuciones tridimensionales de la orientación de la molécula en las tres fases cristalinas diferentes. Cuando se eleva la temperatura (naranja → rojo → amarillo) las moléculas pueden alcanzar más orientaciones. La distribución roja corresponde a la estructura de temperatura ambiente. Crédito:Menno Bokdam / Universidad de Viena