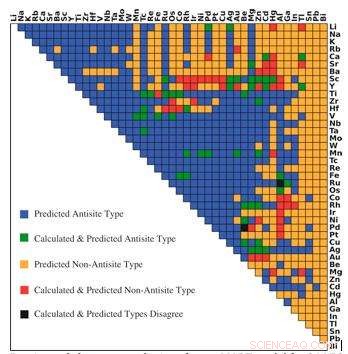
Predicciones del tipo de defecto dominante del modelo r-MART para 946 intermetálicos de tipo B2. Los colores indican la relación entre la predicción y los cálculos como se muestra en la leyenda. Crédito:Bharat Medasani, Laboratorio de Berkeley / PNNL
Por primera vez, Los investigadores del Laboratorio Nacional Lawrence Berkeley (Berkeley Lab) han construido y entrenado algoritmos de aprendizaje automático para predecir el comportamiento de defectos en ciertos compuestos intermetálicos con alta precisión. Este método acelerará la investigación de nuevas aleaciones avanzadas y nuevos materiales ligeros para aplicaciones que abarcan desde la automoción hasta la industria aeroespacial y mucho más.
Sus resultados fueron publicados en la edición de diciembre de 2016 de Materiales computacionales de la naturaleza .
Los materiales nunca son químicamente puros y estructuralmente impecables. Casi siempre contienen defectos, que juegan un papel importante en el dictado de propiedades. Estos defectos pueden aparecer como vacantes, que son esencialmente 'agujeros' en la estructura cristalina de la sustancia, o defectos antisitio, que son esencialmente átomos colocados en el lugar equivocado del cristal. La comprensión de tales defectos puntuales es crucial para los científicos que diseñan materiales porque pueden tener un efecto dramático en la estabilidad y resistencia estructural a largo plazo.
Tradicionalmente, Los investigadores han utilizado un método mecánico cuántico computacional conocido como cálculos funcionales de densidad para predecir qué tipos de defectos se pueden formar en una estructura determinada y cómo afectan las propiedades del material. Aunque eficaz, este enfoque es muy costoso desde el punto de vista computacional de ejecutar para defectos puntuales que limitan el alcance de tales investigaciones.
"Los cálculos funcionales de densidad funcionan bien si está modelando una unidad pequeña, pero si desea hacer que su celda de modelado sea más grande, la potencia computacional requerida para hacer esto aumenta sustancialmente, "dice Bharat Medasani, ex postdoctorado de Berkeley Lab y autor principal del artículo de npj. "Y debido a que es computacionalmente costoso modelar defectos en un solo material, hacer este tipo de modelado de fuerza bruta para decenas de miles de materiales no es factible ".
Para superar estos desafíos informáticos, Medasani y sus colegas desarrollaron y entrenaron algoritmos de aprendizaje automático para predecir defectos puntuales en compuestos intermetálicos, centrándose en la estructura cristalina B2 ampliamente observada. Inicialmente, seleccionaron una muestra de 100 de estos compuestos de la base de datos de proyectos de materiales y realizaron cálculos funcionales de densidad en supercomputadoras en el Centro Nacional de Computación Científica de Investigación Energética (NERSC), una instalación para usuarios de la Oficina de Ciencias del DOE en Berkeley Lab, para identificar sus defectos.
Debido a que tenían una pequeña muestra de datos para trabajar, Medasani y su equipo utilizaron un enfoque forestal llamado aumento de gradiente para desarrollar su método de aprendizaje automático con una alta precisión. En este enfoque, se construyeron sucesivamente modelos adicionales de aprendizaje automático y se combinaron con modelos anteriores para minimizar la diferencia entre las predicciones de los modelos y los resultados de los cálculos funcionales de densidad. Los investigadores repitieron el proceso hasta que lograron un alto nivel de precisión en sus predicciones.
"Este trabajo es esencialmente una prueba de concepto. Muestra que podemos ejecutar cálculos funcionales de densidad para unos pocos cientos de materiales, luego, entrene algoritmos de aprendizaje automático para predecir con precisión defectos puntuales para un grupo mucho más grande de materiales, "dice Medasani, quien ahora es investigador postdoctoral en el Laboratorio Nacional del Noroeste del Pacífico.
"El beneficio de este trabajo es que ahora tenemos un enfoque de aprendizaje automático computacionalmente económico que puede predecir de forma rápida y precisa defectos puntuales en nuevos materiales intermetálicos", dice Andrew Canning, un científico computacional del laboratorio de Berkeley y coautor del artículo de npj. "Ya no tenemos que ejecutar cálculos de primer principio muy costosos para identificar las propiedades de los defectos para cada nuevo compuesto metálico".
"Esta herramienta nos permite predecir defectos metálicos de forma más rápida y robusta, lo que a su vez acelerará el diseño de materiales, "dice Kristin Persson, un científico del laboratorio de Berkeley y director del proyecto de materiales, una iniciativa destinada a reducir drásticamente el tiempo necesario para inventar nuevos materiales al proporcionar acceso abierto basado en la web a información computada en materiales conocidos y previstos. Como extensión de este trabajo, se ha desarrollado un kit de herramientas Python de código abierto para modelar defectos puntuales en semiconductores y aislantes (PyCDT).