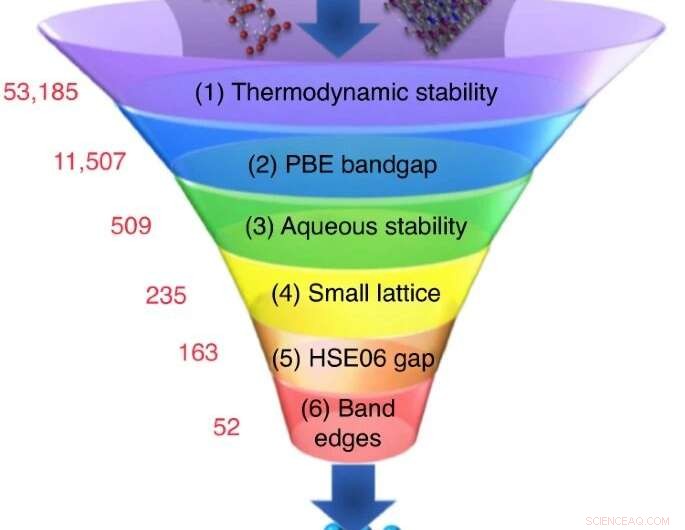
Una gran cantidad de materiales candidatos se eligen de bases de datos experimentales o computacionales, y una secuencia de cálculos de selección reduce su número a un pequeño grupo de candidatos con las propiedades más prometedoras. Crédito:Nicola Marzari
Nicola Marzari, jefe del laboratorio de Teoría y Simulación de Materiales de EFPL y director de NCCR MARVEL, acaba de publicar una revisión de los métodos de estructura electrónica como parte de una edición especial Insight on Computational Materials Design, publicado por Materiales de la naturaleza . El artículo, escrito con Andrea Ferretti de CNR – Instituto Nanoscienze y Chris Wolverton de Northwestern University, proporciona una descripción general de estos métodos, analiza su aplicación a la predicción de las propiedades de los materiales, y examina diferentes estrategias utilizadas para apuntar a los objetivos más amplios del diseño y descubrimiento de materiales. Mirando hacia el futuro, los autores consideran los desafíos emergentes en la precisión predictiva de los cálculos, y abordar la complejidad de la vida real de materiales y dispositivos. También destacan la importancia de las infraestructuras computacionales que sustentan dicha investigación, y cómo la planificación para financiarlos y los modelos profesionales de apoyo apenas está comenzando a surgir.
Durante los últimos 20 años, Las simulaciones de primeros principios se han vuelto poderosas, herramientas ampliamente utilizadas en muchos, diversos campos de la ciencia y la ingeniería. De la nanotecnología a la ciencia planetaria, de la metalurgia a los materiales cuánticos, han acelerado la identificación, caracterización, y optimización de materiales enormemente. Han llevado a predicciones asombrosas, desde el transporte térmico ultrarrápido hasta la superconductividad mediada por electrones y fonones en los hidruros hasta la aparición de bandas planas en el grafeno de dos capas retorcidas, que han inspirado experimentos notables.
El impulso actual para complementar los experimentos con simulaciones; continuado, rápido crecimiento de la capacidad de rendimiento de las computadoras; La capacidad del aprendizaje automático y la inteligencia artificial para acelerar el descubrimiento de materiales, así como la promesa de aceleradores disruptivos como la computación cuántica para tareas exponencialmente costosas, significa que es evidente que estos métodos serán cada vez más relevantes a medida que pase el tiempo. Es un momento apropiado entonces para revisar las capacidades, así como las limitaciones de los métodos de estructura electrónica subyacentes a estas simulaciones. Marzari, Ferretti y Wolverton abordan esta tarea en el artículo "Métodos de estructura electrónica para el diseño de materiales, "recién publicado en Materiales de la naturaleza .
"Las simulaciones no fallan de manera espectacular, pero pueden cambiar sutilmente de ser invaluables a apenas lo suficientemente buenas a simplemente inútiles, ", dijeron los autores en el artículo." Las razones del fracaso son múltiples, desde ampliar las capacidades de los métodos hasta abandonar la complejidad de los materiales reales. Pero las simulaciones también son insustituibles:pueden evaluar materiales en condiciones de presión y temperatura tan extremas que ningún experimento en la tierra es capaz de replicar. pueden explorar con una agilidad cada vez mayor el vasto espacio de las fases y composiciones de los materiales en la búsqueda de ese escurridizo avance de los materiales, y pueden identificar directamente las causas microscópicas y el origen de una propiedad macroscópica. Último, comparten con todas las ramas de la ciencia computacional un elemento clave de la investigación:pueden volverse reproducibles, abiertos y compartibles de una manera que ninguna infraestructura física lo será jamás ".
Los autores primero analizan el marco de la teoría funcional de la densidad (DFT) y brindan una descripción general de los enfoques cada vez más complejos que pueden mejorar la precisión o ampliar el alcance de las simulaciones. Luego, discuten las capacidades que ha desarrollado la ciencia de los materiales computacionales para explotar esta caja de herramientas y entregar predicciones para las propiedades de los materiales en condiciones realistas de complejidad cada vez mayor. Finalmente, destacan cómo los enfoques basados en la física o los datos pueden proporcionar alto rendimiento o vías de inteligencia artificial para el descubrimiento de materiales, y explicar cómo estos esfuerzos están cambiando todo el ecosistema de investigación.
Mirando hacia el futuro, los autores dicen que desarrollar métodos que puedan evaluar la estabilidad termodinámica, condiciones de síntesis, fabricabilidad, y la tolerancia de las propiedades predichas a defectos intrínsecos y extrínsecos en materiales nuevos será un desafío significativo. Los investigadores pueden necesitar aumentar las estimaciones de DFT mediante métodos de estructura electrónica más avanzados o algoritmos de aprendizaje automático para mejorar la precisión. y utilizar métodos computacionales para abordar condiciones realistas como entropías vibratorias, la concentración de defectos y potenciales electroquímicos aplicados.
Finalmente, Dado el papel extendido que probablemente desempeñen estos métodos en las próximas décadas, los autores señalan que el apoyo y la planificación de las infraestructuras computacionales necesarias:software científico ampliamente utilizado, la verificación de códigos y validación de teorías, la difusión y conservación de datos computacionales, las herramientas y los flujos de trabajo, así como los modelos de carrera asociados que estos implican y requieren, apenas están comenzando a emerger.