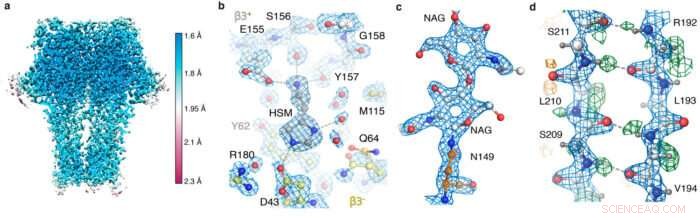
GABA A instantáneas del mapa del receptor. (a) resolución local; (b) la bolsa de agonista que muestra la coordinación de la histamina y las moléculas de agua; (c) glicano ligado a N; (d) red de enlaces de hidrógeno revelada por el mapa de diferencias (picos verdes).
Observar la disposición tridimensional precisa de los átomos dentro de una proteína nos ayuda a comprender cómo puede realizar sus funciones. Aunque la crio-microscopía electrónica (crio-EM) se ha desarrollado rápidamente como una importante técnica de biología estructural en los últimos años, La cristalografía de rayos X había sido la única técnica capaz de visualizar átomos individuales. Los grupos de Radu Aricescu y Sjors Scheres en el Laboratorio de Biología Molecular del MRC, en colaboración con científicos de Thermo Fisher Scientific y otros lugares, ahora han podido resolver átomos de proteínas individuales por primera vez en una imagen crio-EM tridimensional.
Esta colaboración comenzó a principios de 2019 cuando Radu y Abhay Kotecha, investigador de Thermo Fisher Scientific, quería probar nuevo hardware crio-EM en una pequeña muestra de proteína de membrana. Receptores GABAA, un enfoque de la investigación de Radu durante más de una década, fueron elegidos porque la resolución más alta alcanzable utilizando la mejor tecnología disponible parecía haber alcanzado un límite de alrededor de 2,5 Ångströms (Å), pero claramente se necesitaba una resolución más alta para un mejor diseño de fármacos.
¿Qué es la resolución atómica?
La resolución generalmente se informa en Ångströms, una unidad de longitud que es una diez mil millonésima parte de un metro o 0,1 nanómetros, y se refiere a la distancia más pequeña entre la que se puede ver que dos objetos están separados.
La longitud de un enlace carbono-carbono típico es de 1,5 Å; otros enlaces en las proteínas son un poco más cortos. Por lo tanto, a medida que la resolución desciende a 1,2 Å, se hace posible ver átomos individuales dentro de una proteína, logrando una verdadera resolución atómica.
Mientras probaba nuevos desarrollos de hardware que incluían una fuente de electrones de pistola de emisión de campo frío, un nuevo filtro de energía, y una cámara nueva, el equipo también tuvo que desarrollar nuevas estrategias de procesamiento. Algoritmos para la corrección de aberraciones ópticas que fueron previamente desarrollados por Jasenko Zivanov en el grupo de Sjors, así como un algoritmo propuesto por Chris Russo y Richard Henderson, jugó un papel crucial en exprimir la mayor parte de la información de las imágenes.
Después de recibir imágenes recopiladas en el nuevo hardware del microscopio por Abhay Kotecha en Thermo Fisher Scientific en Eindhoven, Países Bajos, Takanori Nakane, un postdoctorado en el grupo de Sjors, desarrolló un flujo de trabajo óptimo en RELION y Andrija Sente, junto con otros miembros del grupo de Radu, utilizó este flujo de trabajo para procesar imágenes del receptor GABAA, mientras retroalimenta los resultados para optimizar rápidamente la configuración del microscopio. Un nuevo El sistema de almacenamiento de datos de alta capacidad desarrollado por Jake Grimmett y Toby Darling en el equipo de Computación Científica de LMB ofreció un soporte crucial para manejar los aproximadamente cien terabytes de datos generados. Este esfuerzo de equipo sostenido condujo a una estructura de receptor GABAA de resolución de 1,7 Å sin precedentes.
Esta fue la resolución mejor reportada lograda usando crio-EM para cualquier muestra de proteína que no sea la proteína apoferritina. La apoferritina se utiliza comúnmente como punto de referencia para crio-EM, porque su estabilidad molecular y simetría de 24 veces permiten reconstrucciones de alta resolución a partir de relativamente pocas partículas.
Usando el nuevo hardware y estrategias de procesamiento, el equipo pudo obtener una estructura de apoferritina de resolución de 1,22 Å, batiendo el récord anterior de 1,53 Å para ser la estructura crio-EM de partícula única de mayor resolución obtenida hasta ahora. Lo más impresionante, esta resolución permitió la visualización de átomos de hidrógeno individuales, incluso en moléculas de agua dentro de la estructura de la proteína. La visualización de las redes de enlaces de hidrógeno dentro de las estructuras de las proteínas y en los bolsillos de unión a los fármacos permite a los investigadores comprender mejor cómo funcionan.
Este trabajo representa la ruptura de una barrera clave para la crio-EM como técnica de biología estructural y la nueva tecnología, recopilación de datos, y las estrategias de procesamiento ampliarán el número de proteínas cuyas estructuras se pueden resolver a alta resolución. Estas reconstrucciones de mayor resolución permitirán una mejor comprensión de cómo funcionan las proteínas y facilitarán el diseño de medicamentos más específicos que podrían tener un impacto en los tratamientos para una amplia gama de enfermedades.