Stuart Lindsay es el director del Centro de Biofísica de Moléculas Únicas en el Instituto de Biodiseño de la Universidad Estatal de Arizona Arizona. Crédito:Instituto de Biodiseño de la Universidad Estatal de Arizona
Unos tres mil millones de pares de bases componen el genoma humano:el plano de la vida. En 2003, el Proyecto Genoma Humano anunció el exitoso descifrado de este código, un tour de force que continúa proporcionando un flujo de conocimientos relevantes para la salud y las enfermedades humanas.
Sin embargo, los actores principales en prácticamente todos los procesos de la vida son las proteínas codificadas por secuencias de ADN conocidas como genes. Para un amplio espectro de enfermedades, Las proteínas pueden producir revelaciones mucho más convincentes de las que se pueden obtener a partir del ADN solo, si los investigadores logran desbloquear las secuencias de aminoácidos de las que están compuestos.
Ahora, Stuart Lindsay y sus colegas del Instituto de Biodiseño de la Universidad Estatal de Arizona han dado un paso importante en esta dirección, demostrando la identificación precisa de aminoácidos, fijando brevemente cada uno en una unión estrecha entre un par de electrodos flanqueantes y midiendo una cadena característica de picos de corriente que pasan a través de moléculas de aminoácidos sucesivas.
Mediante el uso de un algoritmo de aprendizaje automático, Lindsay y su equipo pudieron entrenar una computadora para reconocer ráfagas de actividad eléctrica que representan la unión momentánea de un aminoácido dentro de la unión. Se demostró que las señales de ruido actúan como huellas digitales fiables, identificación de aminoácidos, incluyendo variantes sutilmente modificadas.
Las proteínas ya brindan una gran cantidad de información pertinente a enfermedades como el cáncer, diabetes y trastornos neurológicos como el Alzheimer, además de proporcionar información clave sobre otro proceso mediado por proteínas:el envejecimiento.
El nuevo trabajo avanza la perspectiva de la secuenciación clínica de proteínas y el descubrimiento de nuevos biomarcadores:balizas de alerta temprana que señalan enfermedades. Más lejos, la secuenciación de proteínas puede transformar radicalmente el tratamiento del paciente, permitiendo un seguimiento preciso de la respuesta de la enfermedad a la terapéutica, a nivel molecular.
Los resultados de la investigación del grupo se informan en la edición avanzada en línea de la revista. Nanotecnología de la naturaleza .
Del genoma al proteoma
Una enorme biblioteca de proteínas, conocida como proteoma, ocupa un lugar central en prácticamente todos los procesos de la vida. Las proteínas son vitales para el crecimiento celular, diferenciación y reparación; catalizan reacciones químicas y brindan defensa contra enfermedades, entre una miríada de funciones de limpieza.
Una de las sorpresas más extrañas que surgieron del Proyecto Genoma Humano es el hecho de que solo alrededor del 1,5 por ciento del genoma codifica proteínas. El resto de los nucleótidos del ADN forman secuencias reguladoras, genes de ARN no codificantes, intrones, y ADN no codificante, (una vez etiquetado burlonamente como "ADN basura"). Esto deja a los humanos con escasos 20-25, 000 genes, un descubrimiento aleccionador dado que el humilde gusano redondo tiene aproximadamente el mismo número. Como señala la profesora Lindsay, la noticia empeora:"Una planta de lirio tiene aproximadamente un orden de magnitud más genes que nosotros, " él dice.
El misterio de organismos complejos como los humanos que tienen un número de genes terriblemente bajo tiene que ver con el hecho de que las proteínas generadas a partir del plano del ADN pueden modificarse de varias maneras. De hecho, los científicos ya han identificado más de 100, 000 proteínas humanas e investigadores como Lindsay creen que esto puede ser solo la punta del iceberg.
Así como las oraciones pueden tener su significado alterado a través de cambios en el orden de las palabras o la puntuación, las proteínas generadas a partir de plantillas de genes pueden cambiar la función (o, a veces, volverse inoperables), a menudo con graves consecuencias para la salud humana. Dos procesos clave que modifican las proteínas se conocen como empalme alternativo y modificación postraduccional. Son los impulsores de la extraordinaria variación de proteínas observada.
El empalme alternativo ocurre cuando se codifican regiones de ARN, (conocidos como exones) se empalman y las regiones no codificantes (conocidas como intrones) se cortan, antes de la traducción en proteínas. Este proceso no siempre ocurre de manera ordenada, con la introducción ocasional de superposiciones de exones o intrones, produciendo proteínas empalmadas alternativamente, cuya función puede verse alterada.
Las modificaciones postraduccionales son marcadores que se agregan después de que se han elaborado las proteínas. Hay muchas formas de modificación postraduccional, incluyendo metilación y fosforilación. Algunas proteínas alteradas realizan funciones vitales, mientras que otros pueden ser aberrantes y estar asociados con enfermedades (o propensión a enfermedades). Varios cánceres están asociados con tales errores de proteínas, que ya se utilizan como marcadores de diagnóstico. Sin embargo, la identificación adecuada de tales proteínas sigue siendo un gran desafío en biomedicina.
Nuevas secuencias
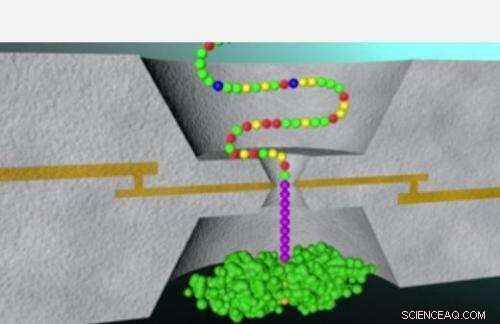
La técnica descrita en la investigación actual se aplicó anteriormente en el laboratorio de Lindsay para la secuenciación exitosa de bases de ADN. Este método, conocido como túnel de reconocimiento, implica enhebrar un péptido a través de un pequeño ojal conocido como nanoporo. Un par de electrodos metálicos, separados por un espacio de aproximadamente dos nanómetros, se asienta a cada lado del nanoporo mientras unidades sucesivas de un péptido pasan a través de la pequeña abertura, con cada unidad completando un circuito eléctrico y emitiendo una ráfaga de picos de corriente.
El grupo de investigación demostró que un análisis detallado de estos picos actuales podría permitir a los investigadores determinar cuál de las cuatro bases de nucleótidos:adenina, timina, citosina o guanina — se colocó entre los electrodos en el nanoporo.
"Hace aproximadamente 2 años, en una de nuestras reuniones de laboratorio, se sugirió que tal vez la misma tecnología funcionaría para los aminoácidos, "Dice Lindsay. Así comenzaron los esfuerzos para abordar el desafío sustancialmente mayor de usar el túnel de reconocimiento para identificar los 20 aminoácidos que se encuentran en las proteínas, a diferencia de solo 4 bases que comprenden ADN.
La secuenciación de una sola molécula de proteínas es de enorme valor, offering the potential to detect diminishingly small quantities of proteins that may have been tweaked by alternative splicing or post-translational modification. A menudo, these are the very proteins of interest from the standpoint of recognizing disease states, though current technologies are inadequate to detect them.
As Lindsay notes, there is no equivalent in the protein world to polymerase chain reaction (PCR) technology, which allows minute quantities of DNA in a sample to be rapidly amplified. "We probably don't even know about most of the proteins that would be important in diagnostics. It's just a black hole to us because the concentrations are too low for current analytical techniques, " él dice, adding that the ability of recognition tunneling to pinpoint abnormalities on a single molecule basis "could be a complete game changer in proteomics."
The new paper describes a series of experiments in which pure samples of individual amino acids, individual molecules in mixed solution and finally, short peptide chains were successfully identified through recognition tunneling. The work sets the stage for a method to sequence individual protein molecules rapidly and cheaply (see accompanying animation).
A machine learning algorithm known as Support Vector Machine was used to train a computer to analyze the burst signals produced when amino acids formed bonds in the tunnel junction and emitted a lively noise signal as the poised electrodes passed tunneling current through each molecule. (The machine learning algorithm is the same one used by the IBM computer 'Watson' to defeat a human opponent in Jeopardy.)
Lindsay says that around 50 distinct signal burst characteristics were used in the amino acid identifications, but that most of the discriminatory power is achieved with 10 or fewer signal traits.
Remarkably, recognition tunneling not only pinpointed amino acids with high reliability from single complex burst signals, but managed to distinguish a post-translationally modified protein (sarcosine) from its unmodified precursor (glycine) and also to discriminate between mirror-image molecules knows as enantiomers and so-called isobaric molecules, which differ in peptide sequence but exhibit identical masses.
Pathway to the $1000 dollar proteome?
Lindsay indicates that the new studies, which rely on innovative strategies for handling single molecules coupled with startling advances in computing power, open up horizons that were inconceivable only a short time ago. It is becoming clear that the tools that made the $1000 genome feasible are equally applicable to an eventual $1000 dollar proteome. En efecto, such a landmark may not be far off. "¿Por qué no?" Lindsay asks. "People think it's crazy but the technical tools are there and what will work for DNA sequencing will work for protein sequencing."
While the tunneling measurements have until now been made using a complex laboratory instrument known as a scanning tunneling microscope (STM), Lindsay and his colleagues are currently working on a solid state device capable of fast, cost-effective and clinically applicable recognition tunneling of amino acids and other analytes. Eventual application of such solid-state devices in massively parallel systems should make clinical proteomics a practical reality.