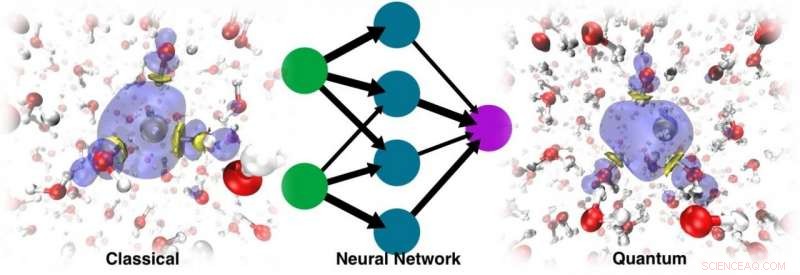
La dinámica ejecutada con el ML PES resultante no solo fue capaz de recuperar la cavidad estable, pero también podría rastrear la dinámica de localización correcta Crédito:@Vladimir Rybkin
El comportamiento del electrón solvatado e-aq tiene implicaciones fundamentales para la electroquímica, fotoquímica, química de alta energía, así como para la biología —su precursor de desequilibrio es responsable del daño por radiación al ADN— y, comprensiblemente, ha sido el tema de investigación experimental y teórica durante más de 50 años.
Aunque el electrón hidratado parece simple, es el anión más pequeño posible, así como el agente reductor más simple en química, capturar su física es ... difícil. Son de corta duración y se generan en pequeñas cantidades, por lo que son imposibles de concentrar y aislar. Por lo tanto, su estructura es imposible de capturar con observación experimental directa, como métodos de difracción o RMN. El modelado teórico ha resultado ser un desafío.
La teoría funcional de la densidad (DFT) es el método de estructura electrónica más utilizado para estudiar el electrón solvatado y el agua. Sin embargo, los funcionales de densidad estándar sufren un error de deslocalización, haciendo imposible modelar radicales con precisión. El agua pura complica considerablemente las aproximaciones DFT, aunque la elección de las funciones adecuadas puede conducir a resultados aceptables en comparación con los valores y puntos de referencia de estructuras electrónicas de alto nivel que se pueden observar a través de experimentos. También se puede lograr una descripción precisa del agua líquida con métodos de química cuántica de muchos cuerpos, pero son extremadamente caros.
Aunque un reciente avance basado en la dinámica molecular a escala de picosegundos, sin precedentes en cuanto a complejidad y que requiere recursos computacionales en los límites de lo posible, proporcionó un argumento crucial a favor de una estructura de cavidad para e-aq, no dio lugar a otros nuevos conocimientos ni a una descripción estadística completa. La caracterización completa de las propiedades del sistema requiere escalas de tiempo mucho más largas, pero la simulación de núcleos cuánticos a este nivel de la teoría de la estructura electrónica está actualmente más allá del alcance computacional.
La forma moderna de solucionar este problema implica el uso del aprendizaje automático. El entrenamiento de un campo de fuerza ML o una superficie de energía potencial (PES) basado en datos ab initio permite simulaciones MD mucho más largas porque el costo de evaluar tales energías y fuerzas es casi insignificante en comparación con el asociado con los cálculos de estructuras electrónicas. El problema es que el electrón solvatado es una especie atípica. No tiene fórmula atomística, lo que plantea un problema porque los PES de aprendizaje automático funcionan con representaciones atomísticas.
En el artículo "Simulando el fantasma:dinámica cuántica del electrón solvatado, "Vladimir Rybkin, investigador de la Universidad de Zúrich, El estudiante de doctorado Jinggang Lan y la profesora Marcella Iannuzzi combinaron su experiencia en estructura electrónica y electrones solvatados con el conocimiento del profesor de EPFL Michele Ceriotti y su ex Ph.D. estudiantes Venkat Kapil, ahora investigador en la Universidad de Cambridge, y Piero Gasparotto, ahora investigador en Empa, en aprendizaje automático y dinámica cuántica. Ese, con las aportaciones de otros compañeros, resultó en la aplicación del enfoque ML a los datos adquiridos a partir de un método de química cuántica de muchos cuerpos conocido como teoría de perturbación de segundo orden de Møller-Plesset (MP2), un método que da una descripción precisa del agua, de todas formas, sin ningún tratamiento especial del exceso de electrones.
Se sorprendieron al descubrir que el modelo podía conocer la presencia del electrón solvatado como un factor que distorsionaba la estructura del agua líquida pura. La dinámica ejecutada con el ML PES resultante no solo fue capaz de recuperar la cavidad estable, pero también podría rastrear la dinámica de localización correcta, partiendo del exceso de electrones deslocalizado añadido al agua. En el final, ML simuló el electrón como una especie de "partícula fantasma" que no estaba explícitamente presente en el modelo.
Esto permitió a los investigadores lograr una escala de tiempo de varios cientos de picosegundos y recopilar estadísticas confiables al ejecutar muchas trayectorias clásicas computacionalmente baratas y calcular espectros vibracionales. estructuras y difusión. El enfoque ML también les permitió simular el cuántico en lugar de los núcleos clásicos con dinámica molecular de ruta integral (PIMD). Esta técnica es al menos un orden de magnitud computacionalmente más cara que la MD clásica y no se puede llevar a cabo sin ML PES a un alto nivel de teoría de estructura electrónica.
Teniendo en cuenta los efectos cuánticos nucleares, se obtuvieron espectros vibracionales precisos, permitiendo a los investigadores cuantificar el impacto de estos efectos, que ya se ha demostrado que son muy importantes en la dinámica de relajación del electrón en exceso, sobre el electrón hidratado. También reveló una difusión transitoria, un inusual, evento raro que no está presente en el régimen clásico. Si bien la difusión no transitoria del electrón solvatado se logra mediante el intercambio de solvente seguido de un desplazamiento gradual de la 'nube de electrones' o distribución de densidad de espín, La difusión transitoria es más bien un salto de la densidad de espín desde la cavidad estable a la adyacente.
Si bien el enfoque de partícula fantasma se aplicó aquí al electrón solvatado, También podría aplicarse a estados excitados y cuasipartículas como polarones, abriendo nuevas oportunidades para unir la teoría de la estructura electrónica de alto nivel con el aprendizaje automático para lograr simulaciones dinámicas de alta precisión a un precio moderado.